Казначейские счета
График закрытия банковских счетов Справочник казначейских счетовСправочник казначейских счетов размещается в виде набора открытых данных.
Перед использованием ознакомьтесь с описанием алгоритма получения и порядка работы со справочником.
Справочник публикуется на ежедневной основе в соответствии с пунктом 3.3 Правил организации и функционирования системы казначейских платежей, утвержденных приказом Федерального казначейства от 13.05.2020 № 20н.
Таблица соответствия счетовТаблица соответствия действующих банковских счетов территориальных органов Федерального казначейства банковским счетам, входящим в состав единого казначейского счета, и казначейским счетам
1 февраля 2021, 14:19 (1 марта 2021, 11:31)
 14 Бюджетного кодекса Российской Федерации (в редакции Федерального закона от 27 декабря 2019 года № 479-ФЗ «О внесении изменений в Бюджетный кодекс Российской Федерации в части казначейского обслуживания и системы казначейских платежей») денежные средства бюджетов, денежные средства, поступающие во временное распоряжение получателей бюджетных средств, денежные средства бюджетных и автономных учреждений, денежные средства юридических лиц, не являющихся участниками бюджетного процесса, бюджетными и автономными учреждениями, лицевые счета которым открыты в Казначействе России (финансовом органе субъекта Российской Федерации, муниципального образования), с 1 января 2021 года учитываются на казначейских счетах.
14 Бюджетного кодекса Российской Федерации (в редакции Федерального закона от 27 декабря 2019 года № 479-ФЗ «О внесении изменений в Бюджетный кодекс Российской Федерации в части казначейского обслуживания и системы казначейских платежей») денежные средства бюджетов, денежные средства, поступающие во временное распоряжение получателей бюджетных средств, денежные средства бюджетных и автономных учреждений, денежные средства юридических лиц, не являющихся участниками бюджетного процесса, бюджетными и автономными учреждениями, лицевые счета которым открыты в Казначействе России (финансовом органе субъекта Российской Федерации, муниципального образования), с 1 января 2021 года учитываются на казначейских счетах. Для совершения переводов денежных средств в целях обеспечения осуществления и отражения операций на казначейских счетах, за исключением казначейских счетов для осуществления и отражения операций с денежными средствами Фонда национального благосостояния, территориальным органам Казначейства России в подразделениях Банка России в первый день функционирования платежной системы Банка России 2021 года открываются банковские счета на балансовом счете № 40102 «Единый казначейский счет» в валюте Российской Федерации, входящие в состав единого казначейского счета.
Открытые в настоящее время территориальным органам Казначейства России банковские счета в валюте Российской Федерации (далее – ранее открытые банковские счета) будут закрыты в первом полугодии 2021 года, при этом предусматривается период одновременного функционирования ранее открытых банковских счетов и банковских счетов, входящих в состав единого казначейского счета. В указанный период допускается зачисление денежных средств на ранее открытые банковские счета, устанавливается запрет списания с них денежных средств, а также предусматривается ежедневный перевод остатка денежных средств с ранее открытых банковских счетов на банковские счета, входящие в состав единого казначейского счета.
Казначейские счета открываются в Федеральном казначействе в соответствии с Порядком открытия казначейских счетов, утвержденным приказом Казначейства России от 1 апреля 2020 года № 15н.
Уважаемый пользователь, данный сайт производит обработку файлов cookie и пользовательских данных (информацию об ip-адресе, местоположении, типе и версии операционной системы, типе и версии браузера, источнике переадресации на сайт, и сведения об открытых страницах пользователя) в целях улучшения функционирования сайта и проведения статистических исследований.
Продолжая использовать сайт, вы даете согласие на сбор и обработку указанной информации (Статья 6 Федерального закона от 27.07.2006 № 152-ФЗ «Закон о персональных данных»).
Понятно
Что такое кредитное плечо?
Принцип рычага в финансах работает так же, как и в физике — с его помощью можно сделать больше и, главное, легче. Финансовый леверидж (рычаг, кредитное плечо) — это соотношение денег трейдера к общему объёму средств, которыми он торгует. По правовой сущности это услуга брокера, предоставляющая средства, превышающие собственные в несколько раз. Использование кредитного плеча позволяет получить больший доход.
Термин «кредитное плечо» довольно сложен в понимании, поэтому давайте разберёмся подробно.
Плечо финансового рычага даёт возможность трейдеру (то есть частному инвестору) совершать сделки стоимостью гораздо выше, чем его собственный капитал на счете. Давайте рассмотрим простой пример. Вы начинающий инвестор и решили, что не можете потратить на операции с активами на фондовом рынке больше, чем 1000$. Но внезапно вы обнаруживаете неплохую стратегию и для сделки с портфелем бумаг вам нужно 20 000$. Недостающие деньги вы одалживаете у своего брокера — получаете кредитное плечо 1:20. Конечно, брокер просто обязан защитить свои деньги, и он в автоматизированной торговой системе выставляет порог для убытка по сделке, равный сумме вашего залога / сумме вашего счёта — 1000$. То есть, если в ходе совершения операций на фондовом рынке вы внезапно понесёте убытки, то они никогда не превысят 1000$ — вы потеряете свои деньги, а брокер, ничем не рискуя, вернёт свои. Это вполне справедливо, и такая ситуация называется margin call — убыточная позиция, угрожающая вам убытками, закрывается (иногда даже без предшествующего уведомления). А если операции на рынке приносят прибыль, то вся прибыль останется вам, а брокер получит обратно только свои кредитные средства. Например, вы купили акции на 20 000$, они стали резко расти, и вы получили аж 3000$ прибыли. Все 3000$ (+1000$ залога) — ваши. Брокер просто заберёт свои 19 000$. Понятно, что, оперируя своими 1000$, вы бы никогда не смогли получить 3000$ прибыли, в этом и заключается основной смысл кредитного плеча. Будьте внимательны: маржин колл касается не только залога, а всей суммы вашего счёта — именно ею вы рискуете, просто в нашем примере эти суммы равны.
А если операции на рынке приносят прибыль, то вся прибыль останется вам, а брокер получит обратно только свои кредитные средства. Например, вы купили акции на 20 000$, они стали резко расти, и вы получили аж 3000$ прибыли. Все 3000$ (+1000$ залога) — ваши. Брокер просто заберёт свои 19 000$. Понятно, что, оперируя своими 1000$, вы бы никогда не смогли получить 3000$ прибыли, в этом и заключается основной смысл кредитного плеча. Будьте внимательны: маржин колл касается не только залога, а всей суммы вашего счёта — именно ею вы рискуете, просто в нашем примере эти суммы равны.
Балансовые счета, виды и характеристики, номер балансового счета физических и юридических лиц, учет собственного имущества предприятия
В бухгалтерском учете балансовые счета — это учетные позиции, показатели которых отражаются в балансе. Все они имеют дебет и кредит. Сумма операций по дебету счёта — дебетовый оборот. Сумма операций по кредиту, — кредитовый оборот. Результат соизмерения оборотов по дебету и кредиту — остаток или сальдо по счёту.
Содержание
Скрыть- Назначение балансовых счетов — учет собственного имущества предприятия.
- Балансовые счета — виды и характеристики
- Балансовые счета организаций
- Балансовые счета физических лиц
- Балансовые счета юридических лиц
- Номер балансового счета
Назначение балансовых счетов — учет собственного имущества предприятия.
Балансовые счета подразделяют на:
— синтетические — счета первого порядка. Их номер обозначается тремя цифрами;
— аналитические — счета второго порядка. Их нумеруют пятью цифрами, причем первые три из них — номер счета первого порядка.
Балансовые счета — виды и характеристики
Основные виды балансовых счетов:
— активные, предназначенные для учета активов предприятия. В них уменьшение средств отражается по кредиту, а увеличение — по дебету, а сальдо бывает только дебетовым;
— пассивные, используемые для учета источников пополнения активов компании. На них уменьшение источников записывается по дебету, а увеличение — по кредиту. Их сальдо всегда будет кредитовым;
На них уменьшение источников записывается по дебету, а увеличение — по кредиту. Их сальдо всегда будет кредитовым;
— активно-пассивные, имеющие свойства двух предыдущих разновидностей счетов.
Существуют и другие виды балансовых счетов: основные, контрарные, регулирующие, дополнительные. Все они предназначены для уточнения расчетов.
Балансовые счета организаций
В банках на балансовых счетах организаций ведутся учетные операции с активами, поступающими от предприятий, учреждений. Так, на счетах первого порядка 10203 и 10204 учитываются акции финансового учреждения, принадлежащие, соответственно, государственным предприятиям и организациям и негосударственным организациям. В разделах 402-409 отражаются операции по обслуживанию клиентов банка, с глубокой детализацией по видам активов, по типам организаций, конкретно по каждому предприятию.
Балансовые счета физических лиц
На балансовых счетах физических лиц финансовые учреждения учитывают операции, проводимые с документами или денежными средствами физических лиц.
Балансовые счета юридических лиц
Согласно плану счетов бухгалтерского учета, финансовые учреждения отражают учетные операции, проводимые с активами, принадлежащими учреждениям, компаниям на балансовых счетах юридических лиц. В качестве примера можно привести счета 61140, 61164, 40802 или 410-407. Именно на них учитывают переводы иностранной валюты, движение по лицевым счетам и другие операции. Для каждого клиента банка, для каждого типа операций выполняется детализация. Порядок формирования номеров счетов второго порядка и структура счетов должны подчиняться общим принципам.
Номер балансового счета
Номера балансовых счетов определяются планом счетов бухгалтерского учета и имеют установленное обозначение, структуру, определенное количество субсчетов, назначение.
Совет от Сравни.ру: Для безошибочного отнесения операций на соответствующий счет, стоит воспользоваться разъяснениями к плану счетов.
Как за 20 лет телекоммуникации в России совершили революцию
1999
Группа компаний Владимира Гусинского «Медиа-мост» приобрела ведущие интернет-ресурсы «Реклама.ру», «Анекдот.ру», журнал «Интернет» и др., создав самую крупную медиаимперию в рунете.
2000
Отмена обязательного для каждого абонента разрешения на использование мобильного телефона.
До первой половины 2000 г. любой желающий не мог просто так пользоваться мобильным телефоном: требовалось специальное разрешение от Госсвязьнадзора, которое стоило $4.
2001
Основан русскоязычный раздел «Википедии».
2002
Вступил в силу закон «Об электронной цифровой подписи», в соответствии с которым электронная цифровая подпись в электронных документах была признана равнозначной собственноручной подписи на бумажных носителях.
2003
В ноябре 2003 г. был принят новый закон «О связи», в 2004 г. он вступит в силу: учрежден фонд, отчисления в который обязаны платить все операторы связи – резерв, средства из которого должны пойти на развитие связи в тех регионах, где это экономически невыгодно.
2004
Rambler Media Ltd. разместила на альтернативной площадке Лондонской фондовой биржи (AIM) 3 млн акций новой эмиссии и больше 800 000 акций существующих акционеров. Компания привлекла $40 млн.
2006
В марте веб-разработчик Альберт Попков запустил проект «Одноклассники», а в октябре Павел Дуров – социальную сеть «В контакте».
2007
Государственная комиссия по радиочастотам выделила сотовым операторам «большой тройки» частоты для оказания услуг мобильной связи 3G.
2008
В России поступил в продажу первый официальный iPhone 3G – и начался бурный рост спроса на смартфоны.
2009
По данным исследования «Яндекса», к концу 2009 г. месячная аудитория рунета достигла 39,7 млн человек (34% населения).
2010
Mail.ru Group разместила акции на Лондонской фондовой бирже. Общая выручка составила $1,003 млрд, $92 млн из которых получила сама Mail.ru Group.
2011
Компания «Скартел» (бренд Yota) запустила в Новосибирске первую в России сеть четвертого поколения – по технологии LTE.
2012
В соответствии с федеральным законом № 39-ФЗ от 28 июля 2012 г. создан единый реестр запрещенных сайтов. Принятие закона сопровождалось забастовкой российского сегмента «Википедии».
2013
Сотовые абоненты получили право менять сотового оператора с сохранением за ними мобильного номера. С тех пор этой возможностью абоненты воспользовались примерно 14 400 000 раз.
2014
Mail.ru Group полностью консолидировала социальную сеть «В контакте». Основатель «В контакте» Павел Дуров покинул Россию.
2015
Московский городской суд принял решение о блокировке Rutracker.org – крупнейшего пиратского торрент-трекера в России.
2016
Президент России Владимир Путин подписал закон Ирины Яровой–Виктора Озерова: все разговоры, сообщения и интернет-активность пользователей должны записываться и храниться полгода-год. Издержки установки оборудования отслеживания и записи разговоров и сообщений законодатель переложил на сотовых операторов и интернет-провайдеров.
2018
Роскомнадзор по постановлению Таганского суда начал блокировать мессенджер Telegram – компания не выполнила требования ФСБ передать ей ключи шифрования: информацию, используемую криптографическим алгоритмом при шифровке – дешифровке сообщений. Заблокировать Telegram не удалось: мессенджер начал использовать ресурсы облачных хостинг-провайдеров Amazon, Google и Microsoft.
featureCounts: эффективная программа общего назначения для сопоставления считываний последовательностей с геномными функциями | Биоинформатика
Аннотация
Мотивация: Технологии секвенирования нового поколения генерируют миллионы прочтений коротких последовательностей, которые обычно выровнены с эталонным геномом. Во многих приложениях ключевой информацией, необходимой для последующего анализа, является количество прочтений, сопоставленных с каждым геномным признаком, например, с каждым экзоном или каждым геном.Процесс подсчета прочтений называется суммированием прочтений. Обобщение прочитанного требуется для большого разнообразия геномных анализов, но до сих пор ему уделялось относительно мало внимания в литературе.
Результаты: Мы представляем featureCounts , программу суммирования прочтений, подходящую для подсчета прочтений, полученных в результате экспериментов по секвенированию РНК или геномной ДНК. featureCounts реализует высокоэффективные методы хэширования хромосом и блокировки признаков.Это значительно быстрее существующих методов (на порядок для суммирования на уровне генов) и требует гораздо меньше компьютерной памяти. Он работает как с одиночными, так и с парными считываниями и предоставляет широкий спектр опций, подходящих для различных приложений секвенирования.
featureCounts реализует высокоэффективные методы хэширования хромосом и блокировки признаков.Это значительно быстрее существующих методов (на порядок для суммирования на уровне генов) и требует гораздо меньше компьютерной памяти. Он работает как с одиночными, так и с парными считываниями и предоставляет широкий спектр опций, подходящих для различных приложений секвенирования.
Доступность и реализация: featureCounts доступен по Стандартной общественной лицензии GNU как часть Subread (http://subread.sourceforge.net) или Rsubread (http://www.bioconductor.org) программные пакеты.
Контактное лицо: [email protected]
1 ВВЕДЕНИЕ
Технологии секвенирования следующего поколения (next-gen) революционизируют биологию, предоставляя возможность секвенировать ДНК с беспрецедентной скоростью (Metzker, 2009; Schuster, 2008). В последние несколько лет вычислительной проблеме сопоставления ридов коротких последовательностей с эталонным геномом уделяется огромное внимание (Fonseca et al. , 2012; Langmead et al., 2009 г.; Ли и Дурбин, 2009 г.; Ляо
, 2012; Langmead et al., 2009 г.; Ли и Дурбин, 2009 г.; Ляо  , 2010).
, 2010).
Несмотря на свою важность в геномных исследованиях, проблеме подсчета прочтений уделялось мало внимания в литературе. Задача может показаться на первый взгляд простой, но на практике имеет много тонкостей.Программы подсчета прочтений должны поддерживать как секвенирование ДНК, так и РНК, а также чтение с одного и парных концов. Подсчитываемые считывания или фрагменты с парными концами могут включать вставки, делеции или слияния по отношению к эталонному геному, и эти сложности необходимо учитывать при сравнении расположения каждого считывания или фрагмента с каждой возможной геномной особенностью-мишенью. Когда количество признаков велико, вычислительная стоимость подсчета считываний может быть сравнима с затратами на шаг выравнивания считываний.
Чтение последовательностей ДНК происходит с помощью различных технологий, включая ChIP-seq для сайтов связывания факторов транскрипции (Valouev et al. , 2008), ChIP-seq для гистоновых меток (Park, 2009) и анализы, выявляющие метилирование ДНК (Harris). и др. , 2010). Геномные особенности, представляющие интерес для считывания ДНК, обычно могут быть указаны в терминах простых геномных интервалов. Например, Pal и др. (2013) подсчитывали чтения, связанные с гистоновыми метками, по промоторным областям генов и по целым генным телам.Росс-Иннес и др. (2012) подсчитали чтения, перекрывающиеся с интервалами, идентифицированными пиковым вызывающим абонентом (Zhang et al. , 2008).
и др. , 2010). Геномные особенности, представляющие интерес для считывания ДНК, обычно могут быть указаны в терминах простых геномных интервалов. Например, Pal и др. (2013) подсчитывали чтения, связанные с гистоновыми метками, по промоторным областям генов и по целым генным телам.Росс-Иннес и др. (2012) подсчитали чтения, перекрывающиеся с интервалами, идентифицированными пиковым вызывающим абонентом (Zhang et al. , 2008).
Подсчет прочтений RNA-seq несколько сложнее из-за необходимости учета сплайсинга экзонов. Один из способов заключается в подсчете прочтений, перекрывающих каждый аннотированный экзон, подход, который можно использовать для проверки альтернативного сплайсинга между экспериментальными условиями (Anders et al. , 2012; Reyes et al. , 2013). Другой распространенный подход заключается в суммировании подсчетов на генном уровне путем подсчета всех прочтений, перекрывающих любой экзон для каждого гена (Anders et al. , 2013; Бхаттачарья и др. , 2013; Человек и др. , 2013). Для этой цели часто используется аннотация генов из RefSeq (Pruitt et al. , 2012) или Ensembl (Flicek et al. , 2012).
, 2013; Бхаттачарья и др. , 2013; Человек и др. , 2013). Для этой цели часто используется аннотация генов из RefSeq (Pruitt et al. , 2012) или Ensembl (Flicek et al. , 2012).
Счетчик прочтений предоставляет общую сводку охвата интересующей геномной особенности. В частности, подсчеты на уровне генов из RNA-seq дают общую сводку об уровне экспрессии гена, но не различают изоформы, когда несколько транскриптов экспрессируются из одного и того же гена.Прочтения обычно могут быть отнесены к генам с хорошей уверенностью, но оценка уровней экспрессии отдельных изоформ по своей природе более сложна, потому что разные изоформы гена обычно имеют высокую долю геномного перекрытия. Был разработан ряд методов, основанных на моделях, которые пытаются деконволюировать уровни экспрессии отдельных транскриптов для каждого гена из данных секвенирования РНК, по существу, используя информацию из прочтений, однозначно отнесенных к областям, где различаются изоформы (Li and Dewey, 2011; Trapnell). и др., 2010). В этой статье основное внимание уделяется проблеме подсчета прочтений, которая обычно применима, даже когда глубина секвенирования недостаточна для надежного анализа на уровне транскриптов. Было разработано множество методов статистического анализа для обнаружения дифференциальной экспрессии или дифференциального связывания на основе количества прочтений (Anders and Huber, 2010; Auer and Doerge, 2011; Hardcastle and Kelly, 2010; Li et al. , 2012; McCarthy ). и др. , 2012; Wu и др. , 2013).Недавние сравнения показали, что методы подсчета прочтений более эффективны по сравнению с методами на основе моделей в целях дифференциальной экспрессии на уровне генов (Nookaew et al. , 2012; Rapaport et al. , 2013) или обнаружения вариаций сплайсинга. (Андерс и др. , 2012).
и др., 2010). В этой статье основное внимание уделяется проблеме подсчета прочтений, которая обычно применима, даже когда глубина секвенирования недостаточна для надежного анализа на уровне транскриптов. Было разработано множество методов статистического анализа для обнаружения дифференциальной экспрессии или дифференциального связывания на основе количества прочтений (Anders and Huber, 2010; Auer and Doerge, 2011; Hardcastle and Kelly, 2010; Li et al. , 2012; McCarthy ). и др. , 2012; Wu и др. , 2013).Недавние сравнения показали, что методы подсчета прочтений более эффективны по сравнению с методами на основе моделей в целях дифференциальной экспрессии на уровне генов (Nookaew et al. , 2012; Rapaport et al. , 2013) или обнаружения вариаций сплайсинга. (Андерс и др. , 2012).
В настоящее время доступно лишь несколько программных средств общего назначения для подсчета прочтений. Пакеты программного обеспечения GenomicRanges (Aboyoun et al. , 2013) и IRanges (Pages et al., 2013), разработанный основной группой проекта Bioconductor (Gentleman et al. , 2004), включают функции для подсчета прочтений, которые перекрывают геномные признаки. Функция countOverlaps из IRanges предназначена для подсчета прочтений, перекрывающихся экзонов или других простых геномных областей, тогда как функция summoverlaps из GenomicRanges предназначена для подсчета прочтений на генном уровне. Другим инструментом является скрипт htseq-count , распространяемый вместе с инфраструктурой HT-Seq Python для обработки данных RNA-seq или DNA-seq (Anders, 2013).Все они являются популярными и хорошо протестированными программными инструментами, но все они широко используют программирование на интерпретируемых компьютерных языках R или Python, и ни один из них не оптимизирован полностью с точки зрения эффективности и скорости. BEDTools — популярный инструмент для поиска перекрытий между геномными признаками, который можно использовать для подсчета перекрытий между прочтениями и признаками (Quinlan and Hall, 2010).
, 2013) и IRanges (Pages et al., 2013), разработанный основной группой проекта Bioconductor (Gentleman et al. , 2004), включают функции для подсчета прочтений, которые перекрывают геномные признаки. Функция countOverlaps из IRanges предназначена для подсчета прочтений, перекрывающихся экзонов или других простых геномных областей, тогда как функция summoverlaps из GenomicRanges предназначена для подсчета прочтений на генном уровне. Другим инструментом является скрипт htseq-count , распространяемый вместе с инфраструктурой HT-Seq Python для обработки данных RNA-seq или DNA-seq (Anders, 2013).Все они являются популярными и хорошо протестированными программными инструментами, но все они широко используют программирование на интерпретируемых компьютерных языках R или Python, и ни один из них не оптимизирован полностью с точки зрения эффективности и скорости. BEDTools — популярный инструмент для поиска перекрытий между геномными признаками, который можно использовать для подсчета перекрытий между прочтениями и признаками (Quinlan and Hall, 2010). Он полностью реализован на компилируемом языке C++, что делает его быстрее, чем вышеупомянутые инструменты. Однако он не предназначен специально для данных RNA-seq, поэтому может подсчитывать считывания только для экзонов или интервальных признаков, аналогично countOverlaps .
Он полностью реализован на компилируемом языке C++, что делает его быстрее, чем вышеупомянутые инструменты. Однако он не предназначен специально для данных RNA-seq, поэтому может подсчитывать считывания только для экзонов или интервальных признаков, аналогично countOverlaps .
В этой статье представлена оптимизированная программа подсчета считываний под названием featureCounts . featureCounts можно использовать для количественной оценки прочтений, полученных с помощью технологий секвенирования РНК или ДНК, с точки зрения любого типа геномных признаков. Он реализует хеширование хромосом, блокировку функций и другие стратегии для назначения операций чтения функциям с высокой эффективностью. Он поддерживает многопоточность, что обеспечивает дальнейшее повышение скорости работы с большими объемами данных. Он доступен либо как команда Unix, либо как функция в пакете R Rsubread .В любом случае все основные функции написаны на языке программирования Си. Функция R — это оболочка для скомпилированного кода C, обеспечивающая удобство среды программирования R без ущерба для эффективности реализации C.
Функция R — это оболочка для скомпилированного кода C, обеспечивающая удобство среды программирования R без ущерба для эффективности реализации C.
2 ФОРМАТЫ ДАННЫХ И ВВОД
2.1 Входные данные
Входные данные для featureCounts состоят из (i) одного или нескольких файлов выровненных чтений в формате Sequence Alignment/Map (SAM) или Binary Alignment/Map (BAM) (Li et al., 2009) и (ii) список геномных признаков либо в формате общих признаков (GFF) (Wellcome Trust Sanger Institute, 2013), либо в упрощенном формате аннотаций (SAF) (Shi and Liao, 2013b). Формат считанного ввода (SAM или BAM) определяется автоматически, поэтому пользователю не нужно указывать его. И выравнивание чтения, и аннотация признаков должны соответствовать одному и тому же эталонному геному, который представляет собой набор эталонных последовательностей, представляющих хромосомы или контиги. Для каждого считывания в файле SAM или BAM указывается имя эталонной хромосомы или контига, с которым сопоставлено считывание, начальная позиция считывания на хромосоме или контиге, а также строка так называемого краткого идиосинкразического отчета о выравнивании с разрывами (CIGAR), содержащая строку подробную информацию о выравнивании, включая вставки и удаления и т.д. относительно начальной позиции.
Для каждого считывания в файле SAM или BAM указывается имя эталонной хромосомы или контига, с которым сопоставлено считывание, начальная позиция считывания на хромосоме или контиге, а также строка так называемого краткого идиосинкразического отчета о выравнивании с разрывами (CIGAR), содержащая строку подробную информацию о выравнивании, включая вставки и удаления и т.д. относительно начальной позиции.
Геномные признаки могут быть указаны в формате GFF или SAF. Формат SAF проще и включает только пять обязательных столбцов для каждого признака: идентификатор признака, название хромосомы, начальное положение, конечное положение и цепь. Эти пять столбцов предоставляют минимально достаточную информацию для целей количественной оценки чтения. В любом формате предполагается, что идентификаторы объектов уникальны в соответствии с широко используемым форматом переноса генов (GTF), уточнением GFF (Brent Lab, 2013).
Количество эталонных последовательностей может быть маленьким или большим в зависимости от приложения. Для хорошо установленных геномов количество эталонных последовательностей равно или близко к количеству хромосом. Однако количество эталонных последовательностей может быть намного больше для геномов с неполными или низкокачественными сборками, потому что каждый контиг становится эталонной последовательностью. Чтения RNA-seq иногда выравниваются с транскриптомом, а не с геномом. В этом случае могут быть сотни тысяч транскриптов, и каждый транскрипт становится эталонной последовательностью.
Для хорошо установленных геномов количество эталонных последовательностей равно или близко к количеству хромосом. Однако количество эталонных последовательностей может быть намного больше для геномов с неполными или низкокачественными сборками, потому что каждый контиг становится эталонной последовательностью. Чтения RNA-seq иногда выравниваются с транскриптомом, а не с геномом. В этом случае могут быть сотни тысяч транскриптов, и каждый транскрипт становится эталонной последовательностью.
featureCounts поддерживает подсчет считываний для конкретных цепей, если предоставлена информация, специфичная для цепей. Результаты сопоставления прочтений обычно включают показатели качества сопоставления для сопоставленных прочтений. Пользователи могут дополнительно указать минимальную оценку качества сопоставления, которой должны удовлетворять назначенные операции чтения.
2.2 Считывания с одинарными и парными концами
Чтения могут быть парными или непарными. Если используются парные чтения, то каждая пара ридов определяет фрагмент ДНК или РНК, объединенный двумя ридами.В этом случае featureCounts будет считать фрагменты, а не чтения. featureCounts автоматически сортирует чтения по имени, если парные чтения не находятся в последовательных позициях в файле SAM или BAM.
Если используются парные чтения, то каждая пара ридов определяет фрагмент ДНК или РНК, объединенный двумя ридами.В этом случае featureCounts будет считать фрагменты, а не чтения. featureCounts автоматически сортирует чтения по имени, если парные чтения не находятся в последовательных позициях в файле SAM или BAM.
2.3 Функции и метафункции
Каждый признак представляет собой интервал (диапазон позиций) на одной из эталонных последовательностей. Мы также определяем метапризнак как набор признаков, представляющих интересующую биологическую конструкцию. Например, признаки часто соответствуют экзонам, а метапризнаки — генам.Объекты, имеющие один и тот же идентификатор объекта в аннотации GFF или SAF, считаются принадлежащими одному и тому же метаобъекту. featureCounts может суммировать чтения на уровне функций или метафункций.
3 АЛГОРИТМ
 1″> 3.1 Перекрытие чтений с функциями
1″> 3.1 Перекрытие чтений с функциямиfeatureCounts выполняет точное присвоение считывания путем сравнения положения сопоставления каждого основания в считывании или фрагменте с областью генома, охватываемой каждым признаком.Он учитывает любые пробелы (инсерции, делеции, экзон-экзонные соединения или слияния), которые обнаруживаются в прочтении. Он вызывает попадание, если обнаруживается какое-либо перекрытие (1 п.н. или более) между чтением или фрагментом и функцией.
Столкновение вызывается для метафункции, если чтение или фрагмент перекрывают любой компонент метафункции.
3.2 Несколько перекрытий
Чтение или фрагмент с множественным перекрытием — это чтение, которое перекрывает более одной функции или более одной метафункции при суммировании на уровне метафункции. featureCounts предоставляет пользователям возможность либо исключить чтение с несколькими перекрытиями, либо подсчитать их для каждой перекрывающейся функции. Решение о подсчете этих прочтений часто определяется типом эксперимента. Мы рекомендуем, чтобы считывания или фрагменты, перекрывающие более одного гена, не учитывались для экспериментов RNA-seq, потому что любой отдельный фрагмент должен происходить только от одного из целевых генов, но идентичность истинного целевого гена не может быть определена с уверенностью. С другой стороны, мы рекомендуем подсчитывать множественные перекрывающиеся чтения или фрагменты для большинства экспериментов ChIP-seq, потому что эпигенетические модификации, выведенные из этих прочтений, могут регулировать биологические функции всех их перекрывающихся генов (Pal et al., 2013).
Решение о подсчете этих прочтений часто определяется типом эксперимента. Мы рекомендуем, чтобы считывания или фрагменты, перекрывающие более одного гена, не учитывались для экспериментов RNA-seq, потому что любой отдельный фрагмент должен происходить только от одного из целевых генов, но идентичность истинного целевого гена не может быть определена с уверенностью. С другой стороны, мы рекомендуем подсчитывать множественные перекрывающиеся чтения или фрагменты для большинства экспериментов ChIP-seq, потому что эпигенетические модификации, выведенные из этих прочтений, могут регулировать биологические функции всех их перекрывающихся генов (Pal et al., 2013).
Обратите внимание, что при подсчете на уровне метафункции чтения, перекрывающие несколько функций одной и той же метафункции, всегда учитываются только один раз для этой метафункции, при условии, что нет перекрытия с какой-либо другой метафункции. Например, чтение, охватывающее экзон, будет учитываться только один раз для соответствующего гена, даже если оно перекрывается более чем с одним экзоном.
3.3 Хеширование хромосом
Первым шагом алгоритма featureCounts является создание хэш-таблицы для имен эталонных последовательностей.Это позволяет быстро сопоставлять имена эталонных последовательностей, найденные в файлах SAM, и аннотации GFF. Это особенно полезно при наличии большого количества эталонных последовательностей. После сопоставления прочтений и признаков по эталонной последовательности последующий анализ может проводиться для каждой эталонной последовательности отдельно.
3.4 Геномные ячейки и блоки признаков
После хеширования имен эталонных последовательностей объекты в каждой эталонной последовательности сортируются по их начальным позициям (крайние левые базовые позиции).Затем для каждой эталонной последовательности создается двухуровневая иерархия. Во-первых, эталонная последовательность делится на неперекрывающиеся интервалы по 128 килобайт, и признаки назначаются интервалам в соответствии с их начальными позициями. В каждом бине равное количество последовательных признаков группируется в блоки (рис. 1). Количество блоков в ячейке — это квадратный корень из числа объектов в этой ячейке (округленный до следующего целого числа). Это гарантирует, что количество объектов в блоке почти равно количеству блоков в бине, что является оптимальным параметром для иерархического поиска.
В каждом бине равное количество последовательных признаков группируется в блоки (рис. 1). Количество блоков в ячейке — это квадратный корень из числа объектов в этой ячейке (округленный до следующего целого числа). Это гарантирует, что количество объектов в блоке почти равно количеству блоков в бине, что является оптимальным параметром для иерархического поиска.
Рис. 1.
Геномные ячейки и блоки признаков. Каждая хромосома разделена на бины по 128 т.п.н. Признаки (сплошные линии под хромосомой) назначаются ячейкам в соответствии с их начальными позициями и группируются в блоки (серые прямоугольники) внутри каждой ячейки. Чтения запросов сравниваются с геномными бинами, затем с блоками (пунктирные стрелки) и, наконец, с признаками (сплошные стрелки). Запрос, считанный на рисунке, перекрывается с двумя признаками в первом блоке бина i
Рис.1.
Геномные ячейки и блоки признаков. Каждая хромосома разделена на бины по 128 т.п.н. Признаки (сплошные линии под хромосомой) назначаются ячейкам в соответствии с их начальными позициями и группируются в блоки (серые прямоугольники) внутри каждой ячейки. Чтения запросов сравниваются с геномными бинами, затем с блоками (пунктирные стрелки) и, наконец, с признаками (сплошные стрелки). Запрос, считанный на рисунке, перекрывается с двумя функциями в первом блоке бина i
Чтения запросов сравниваются с геномными бинами, затем с блоками (пунктирные стрелки) и, наконец, с признаками (сплошные стрелки). Запрос, считанный на рисунке, перекрывается с двумя функциями в первом блоке бина i
Использование иерархической структуры данных (функции внутри блоков внутри бинов) является ключевым компонентом алгоритма featureCounts .Это облегчает назначение быстрого чтения, быстро сужая область генома, которая может содержать функции, перекрывающиеся с чтением запроса. Прочитанный запрос сначала сравнивается с бинами геномики, затем с блоками признаков в любых перекрывающихся бинах, а затем с признаками в любых перекрывающихся блоках. Вместо использования нескольких уровней бинов (Кент и др. , 2002; Куинлан и Холл, 2010) алгоритм использует только один уровень бинов в сочетании с функциональными блоками. Наконец, алгоритм решает, как назначить чтение в соответствии с тем, какой уровень суммирования выполняется (уровень функции или уровень метафункции) и разрешено ли чтение перекрываться более чем одной целью на этом уровне.
4 РЕАЛИЗАЦИЯ
Команда featureCounts в пакете Subread для Unix полностью написана на языке программирования C. Объем памяти сводится к минимуму за счет хранения в памяти только данных аннотаций объектов, необходимых на каждом этапе вычислений. Код C поддерживает многопоточность, и пользователь может указать количество используемых потоков. Один поток по умолчанию.
Функция R featureCounts в пакете Rsubread для R является оболочкой для того же скомпилированного кода C, что и для командной строки Unix.Функция R обеспечивает удобство среды программирования R без ущерба для эффективности реализации C. Он создает объект данных в R, который может быть введен непосредственно в программное обеспечение для статистического анализа на основе R, такое как edgeR (Robinson et al. , 2010) или limma (Law et al. , 2013), которые предназначен для анализа количества прочтений следующего поколения.
5 ХАРАКТЕРИСТИКИ НА ДАННЫХ RNA-SEQ
5.1 Данные и аннотация
Сначала мы сравним производительность featureCounts с существующими программными инструментами для подсчета последовательностей РНК на генном уровне. В качестве примера тематического исследования мы используем данные секвенирования РНК, которые были получены в рамках проекта SEQC (контроль качества секвенирования), третьего этапа проекта контроля качества MicroArray (MAQC) (Shi et al. , 2006). Эти данные состоят из 6,8 миллионов пар прочтений длиной 101 п.н., полученных путем секвенирования образца универсальной эталонной РНК человека на приборе Illumina HiSeq 2000.
Набор данных SEQC RNA-seq был сопоставлен с геномом человека GRCh47 с помощью выравнивателя Subjunc , входящего в пакет Subread (Liao and Shi, 2013; Liao et al. , 2013; Shi and Liao, 2013a). . Мы использовали Subjunc для этого анализа, потому что он явно идентифицирует экзон-экзонные соединения и выводит местоположение сопоставления каждой базы каждого чтения, включая те, которые охватывают несколько экзонов. Это позволило нам тщательно изучить способность программ подсчета считываний подсчитывать считывания, охватывающие несколько экзонов, а также считывания, попадающие в экзоны.
, 2013; Shi and Liao, 2013a). . Мы использовали Subjunc для этого анализа, потому что он явно идентифицирует экзон-экзонные соединения и выводит местоположение сопоставления каждой базы каждого чтения, включая те, которые охватывают несколько экзонов. Это позволило нам тщательно изучить способность программ подсчета считываний подсчитывать считывания, охватывающие несколько экзонов, а также считывания, попадающие в экзоны.
Гены и экзоны были определены, как в аннотации NCBI человека RefSeq build 37.2. Это включало 25 702 гена и 225 071 экзон.
Подсчет суммирован на генном уровне. То есть экзоны были определены как признаки, гены были определены как метапризнаки, а количественная оценка была на уровне метапризнаков. Поскольку это данные секвенирования РНК, прочтения или фрагменты, которые перекрываются с несколькими генами, должны быть исключены из подсчета.
5.2 Сравнительная производительность при подсчете чтений
Чтобы продемонстрировать featureCounts при одностороннем чтении, первая оценка использует только первое чтение из каждой пары чтения. В таблице 1 сравнивается производительность featureCounts с производительностью функции summoverlaps пакета GenomicRanges и сценария htseq-count . featureCounts и sumOverlaps дали идентичные подсчеты для каждого гена (таблица 1, столбец 2).
В таблице 1 сравнивается производительность featureCounts с производительностью функции summoverlaps пакета GenomicRanges и сценария htseq-count . featureCounts и sumOverlaps дали идентичные подсчеты для каждого гена (таблица 1, столбец 2).
по данным SEQC RNA-seq
| Метод . | Количество чтений . | Количество фрагментов . | Время (мин) . | Память (МБ) . | |
|---|---|---|---|---|---|
| featureCounts | 4 385 354 | 4 796 948 | 1,0 | 16 | |
| SummarizeOverlaps (весь геном сразу) | 4 385 354 | 3 942 439 | 12.1 | 3400 | |
| SummarizeOverlaps (по хромосомам) | 4 385 354 | 3 942 439 4 | 2 | ||
| 661 | 661 | ||||
| htseq-count 2 | 4 385 4 769 913 | 4 769 913 | 22.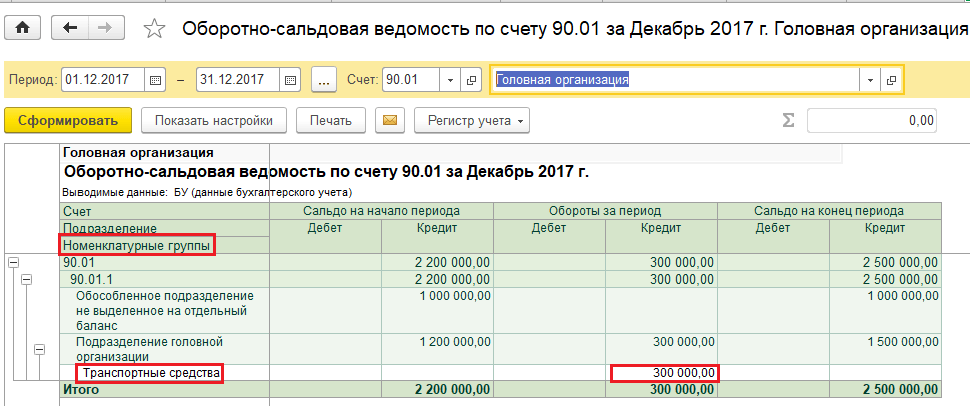 7 7 | 101 | 101 |
| Метод . | Количество чтений . | Количество фрагментов . | Время (мин) . | Память (МБ) . 1.0 | 16 | 16 | |
|---|---|---|---|---|---|---|---|
| Summarizeverlaps (весь генома сразу) | 4 385 354 | 3 942 439 | 12.1 | 39004 | |||
| Summarizeverllaps (Chromosome) | 4 385 354 | 3 942 439 | 41.7 | 661 | 661 | ||
| 4 385 207 | 9 4 769 913 4 769 913 22.7 | 101 | |
Результаты работы с данными SEQC RNA-seq
| Метод . | Количество чтений . | Количество фрагментов . | Время (мин)
. | Память (МБ) . |
|---|---|---|---|---|
| featureCounts | 4 385 354 | 4 796 948 | 1,0 | 16 |
| SummarizeOverlaps (весь геном сразу) | 4 385 354 | 3 942 439 | 12.1 | 3400 |
| SummarizeOverlaps (хромосомой) | 4 385 354 | 3 942 439 | 41,7 | 661 |
| htseq-кол | 4 385 207 | 4 769 913 | 22,7 | 101 |
| Метод . | Количество чтений . | Количество фрагментов . | Время (мин) . | Память (МБ) . | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| featureCounts | 4 385 354 | 4 796 948 | 1,0 | 16 | ||||||||||
| SummarizeOverlaps (весь геном сразу) | 4 385 354 | 3 942 439 | 12.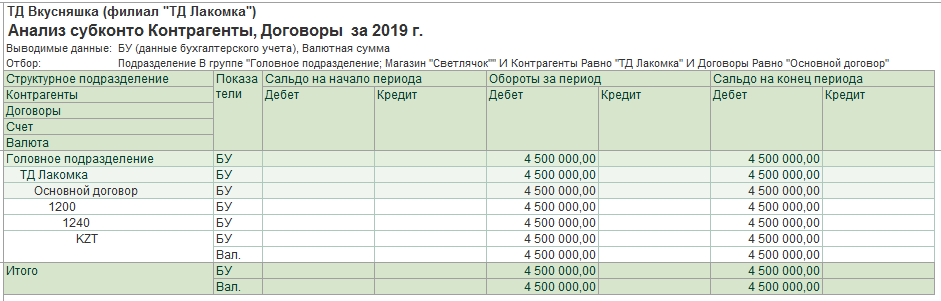 1 1 | 3400 | ||||||||||
| SummarizeOverlaps (по хромосомам) | 4 385 354 | 3 942 439 4 | 2 | |||||||||||
| 661 | 661 | |||||||||||||
| 9002 | | 4 385 4 769 913 | 913 | 22.7 | 101 |
 Спецификация GFF утверждает, что начальное и конечное положения признаков являются инклюзивными (Wellcome Trust Sanger Institute, 2013), поэтому интерпретация featureCounts и sumOverlaps представляется правильной.GFF — единственный формат аннотаций, поддерживаемый htseq-count . Мы изменили файл аннотации, предоставленный для htseq-count , добавив единицу в крайнюю правую позицию каждого экзона, чтобы htseq-count включал эти позиции. После этой модификации htseq-count дал количество, идентичное featureCounts и sumOverlaps .
Спецификация GFF утверждает, что начальное и конечное положения признаков являются инклюзивными (Wellcome Trust Sanger Institute, 2013), поэтому интерпретация featureCounts и sumOverlaps представляется правильной.GFF — единственный формат аннотаций, поддерживаемый htseq-count . Мы изменили файл аннотации, предоставленный для htseq-count , добавив единицу в крайнюю правую позицию каждого экзона, чтобы htseq-count включал эти позиции. После этой модификации htseq-count дал количество, идентичное featureCounts и sumOverlaps . ) или фрагменты ( b ) к генам. Набор данных тот же, что и для таблицы 1.Перекрытие диаграммы Венна дает количество прочтений или фрагментов, присвоенных обоими методами одному и тому же гену. Остальные подсчеты дают количество прочтений или фрагментов, присвоенных одним методом некоторым генам, но не другим методом
) или фрагменты ( b ) к генам. Набор данных тот же, что и для таблицы 1.Перекрытие диаграммы Венна дает количество прочтений или фрагментов, присвоенных обоими методами одному и тому же гену. Остальные подсчеты дают количество прочтений или фрагментов, присвоенных одним методом некоторым генам, но не другим методом Summarize Overlaps насчитал гораздо меньше фрагментов, чем featureCounts и htseq-count (таблица 1, столбец 3).Основная причина этого несоответствия заключается в том, что sumOverlaps требует, чтобы оба конца фрагментов были успешно сопоставлены перед их назначением генам, в то время как featureCounts и htseq-count не имеют такого требования, т. е. они могут назначать фрагменты только один раз. конец отображен. При длине считывания 101 п.н. фрагменты только с одним картированным концом могут иметь относительно высокую достоверность картирования. Подсчет таких фрагментов, вероятно, принесет пользу последующему анализу.Многие элайнеры сообщают о фрагментах, у которых картирован только один конец, включая Subread, Subjunc (Liao et al. , 2013), Bowtie (Langmead et al. , 2009) и TopHat (Trapnell et al. , 2009). Почти все (92%) фрагменты, подсчитанные с помощью featureCounts , но не с помощью summoverlaps , были отнесены к генам, которые также имели по крайней мере 100 присвоенных фрагментов с картированием обоих концов.
Summarize Overlaps насчитал гораздо меньше фрагментов, чем featureCounts и htseq-count (таблица 1, столбец 3).Основная причина этого несоответствия заключается в том, что sumOverlaps требует, чтобы оба конца фрагментов были успешно сопоставлены перед их назначением генам, в то время как featureCounts и htseq-count не имеют такого требования, т. е. они могут назначать фрагменты только один раз. конец отображен. При длине считывания 101 п.н. фрагменты только с одним картированным концом могут иметь относительно высокую достоверность картирования. Подсчет таких фрагментов, вероятно, принесет пользу последующему анализу.Многие элайнеры сообщают о фрагментах, у которых картирован только один конец, включая Subread, Subjunc (Liao et al. , 2013), Bowtie (Langmead et al. , 2009) и TopHat (Trapnell et al. , 2009). Почти все (92%) фрагменты, подсчитанные с помощью featureCounts , но не с помощью summoverlaps , были отнесены к генам, которые также имели по крайней мере 100 присвоенных фрагментов с картированием обоих концов.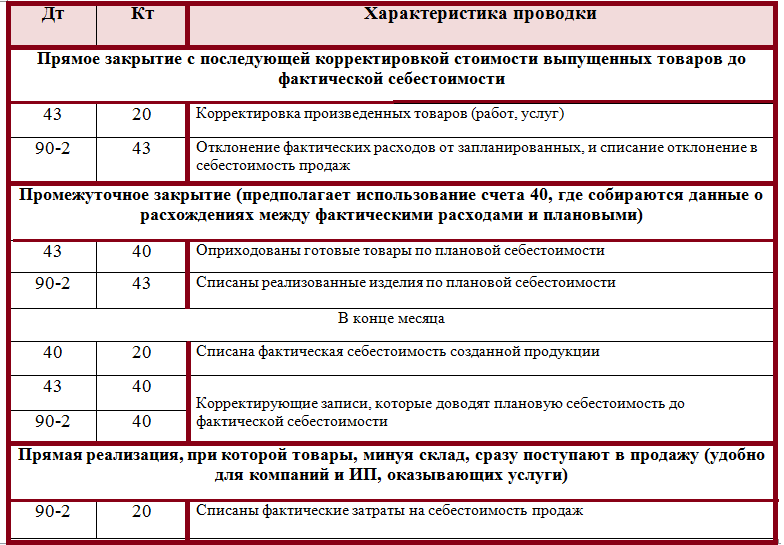 Это показывает, что фрагменты были отнесены к действительно экспрессированным генам, что дает уверенность в том, что дополнительные фрагменты были отнесены правильно.Только 0,1% подсчетов дополнительных фрагментов по featureCounts были отнесены к генам, не поддерживаемым каким-либо фрагментом с картированными обоими концами.
Это показывает, что фрагменты были отнесены к действительно экспрессированным генам, что дает уверенность в том, что дополнительные фрагменты были отнесены правильно.Только 0,1% подсчетов дополнительных фрагментов по featureCounts были отнесены к генам, не поддерживаемым каким-либо фрагментом с картированными обоими концами. Однако htseq-count воспримет этот фрагмент как неоднозначный и не отнесет его ни к одному гену. Это основная причина, по которой featureCounts насчитал немного больше фрагментов, чем htseq-count . featureCounts использует размер перекрытия (с точки зрения чтений) для восстановления этих «неоднозначных» фрагментов.Для этого набора данных >86% фрагментов, присвоенных featureCounts , но не htseq-count , были отнесены к генам, у которых уже было не менее 100 однозначных фрагментов, присвоенных обоими методами. Только 0,2% дополнительных фрагментов, назначенных featureCounts , не поддерживались обычно назначаемыми фрагментами. Это снова показывает, что дополнительные фрагменты относятся к действительно экспрессированным генам, что позволяет предположить, что дополнительные фрагменты, вероятно, были правильно назначены.
Однако htseq-count воспримет этот фрагмент как неоднозначный и не отнесет его ни к одному гену. Это основная причина, по которой featureCounts насчитал немного больше фрагментов, чем htseq-count . featureCounts использует размер перекрытия (с точки зрения чтений) для восстановления этих «неоднозначных» фрагментов.Для этого набора данных >86% фрагментов, присвоенных featureCounts , но не htseq-count , были отнесены к генам, у которых уже было не менее 100 однозначных фрагментов, присвоенных обоими методами. Только 0,2% дополнительных фрагментов, назначенных featureCounts , не поддерживались обычно назначаемыми фрагментами. Это снова показывает, что дополнительные фрагменты относятся к действительно экспрессированным генам, что позволяет предположить, что дополнительные фрагменты, вероятно, были правильно назначены. sumOverlaps также запускался хромосома за хромосомой для экономии памяти. То есть чтения были разделены на группы в соответствии с хромосомами, с которыми они были картированы, и каждая группа прочтений суммировалась отдельно. Но он по-прежнему использовал в 20 раз больше памяти, чем featureCounts .
sumOverlaps также запускался хромосома за хромосомой для экономии памяти. То есть чтения были разделены на группы в соответствии с хромосомами, с которыми они были картированы, и каждая группа прочтений суммировалась отдельно. Но он по-прежнему использовал в 20 раз больше памяти, чем featureCounts . Этот набор данных состоит из 15 миллионов пар считываний ДНК размером 35 п.н., сгенерированных анализатором генома Illumina IIx. В исследовании было проанализировано общее количество фрагментов, картированных в широкой области каждого гена, где широкая область определяется как все тело гена от первого до последнего основания плюс область размером 3 т.п.н. непосредственно выше начала транскрипции гена, представляющего предполагаемая промоторная область (Pal et al. , 2013).
Этот набор данных состоит из 15 миллионов пар считываний ДНК размером 35 п.н., сгенерированных анализатором генома Illumina IIx. В исследовании было проанализировано общее количество фрагментов, картированных в широкой области каждого гена, где широкая область определяется как все тело гена от первого до последнего основания плюс область размером 3 т.п.н. непосредственно выше начала транскрипции гена, представляющего предполагаемая промоторная область (Pal et al. , 2013). 2).
2).| Метод . | Количество фрагментов . | Время (мин)
. | Память (МБ) . | ||
|---|---|---|---|---|---|
| 5 392 155 | 0.9 | 4 | 4 | | |
| Усилители (весь генома сразу) | 5 392 155 | 24.4 | 7000 | 7000 | 9000 |
| Усилители (Chromosome) | 5 392 155 | 36.6 | 783 | ||
| HTSEQ-Count (Союз) | 4 978 050 | 36,0 | 31 | ||
| 4 993 644 | 4 993 644 | 35.7 | 35.7 | 31 | |
| Обложка | 5 366 902 | 4.4 | 41 |
| Метод . | Количество фрагментов . | Время (мин) . | Память (МБ) . | |||||
|---|---|---|---|---|---|---|---|---|
| featureCounts | 5 392 155 | 0,9 | 4 | |||||
| CountOverlaps (весь геном сразу) | 5 392 155 | 24,4 | 7000 | |||||
| CountOverlaps (по хромосоме) | 5 392 155 | 36. 6 6 | 783 | |||||
| | 9 4 978 050 36.0 | 36.0 | 31 | HTSEQ-Count (пересечение) | 4 993 644 | 35.7 | 31 | |
| Обложка | 5 366 902 | 9 441 | 41 |
Выполнение результатов производительности на CHIP-SEQ H4K27ME3
| Метод . | Количество фрагментов . | Время (мин) . | Память (МБ) . | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| featureCounts | 5 392 155 | 0,9 | 4 | |||||||||||
| CountOverlaps (весь геном сразу) | 5 392 155 | 24,4 | 7000 | |||||||||||
| CountOverlaps (по хромосоме) | 5 392 155 | 36.6 | 783 | |||||||||||
| | 9 4 978 050 36. |  0 0 36.0 | 31 | HTSEQ-Count (пересечение) | 4 993 644 | 35.7 | 31 | Охват | 5 366 902 | 49 | 41 | 41 | |
| Метод . | Количество фрагментов . | Время (мин) . | Память (МБ) . | ||
|---|---|---|---|---|---|
| featureCounts | 5 392 155 | 0,9 | 4 | ||
| CountOverlaps (весь геном сразу) | 5 392 155 | 24,4 | 7000 | ||
| CountOverlaps (хромосомой) | 5 392 155 | 5 392 155 | 36,6 | 783 | 783 |
| HTSEQ-Count (Союз) | 4 978 050 | 36.0 | 31 | ||
| htseq-кол- (пересечение-непустое) | 4 993 644 | 35,7 | 31 | ||
| coverageBED | 5 366 902 | 4,4 | 41 |
featureCounts и countOverlaps дали одинаковые подсчеты для каждого гена, но featureCounts был значительно быстрее и эффективнее использовал память. countOverlaps также запускался хромосома за хромосомой для экономии памяти.Это уменьшило пиковое использование памяти, хотя оно оставалось более чем в сто раз больше, чем использовалось featureCounts . Обратите внимание, что featureCounts , в отличие от countOverlaps , может подсчитывать фрагменты с успешно сопоставленным только одним концом, но такие фрагменты не были включены в эту оценку, чтобы гарантировать, что тайминги и использование памяти для featureCounts и countOverlaps были для идентичных операций. .
countOverlaps также запускался хромосома за хромосомой для экономии памяти.Это уменьшило пиковое использование памяти, хотя оно оставалось более чем в сто раз больше, чем использовалось featureCounts . Обратите внимание, что featureCounts , в отличие от countOverlaps , может подсчитывать фрагменты с успешно сопоставленным только одним концом, но такие фрагменты не были включены в эту оценку, чтобы гарантировать, что тайминги и использование памяти для featureCounts и countOverlaps были для идентичных операций. .
coverageBED присвоено немного меньше фрагментов, чем featureCounts .Мы обнаружили, что это произошло из-за того, что покрытие BED использовало только первое чтение каждого фрагмента, чтобы присвоить весь фрагмент функциям. htseq-count насчитал на 7–8% меньше фрагментов, предположительно, потому, что он не подсчитывает фрагменты с множественным перекрытием. htseq-count запускался в режиме «пересечение-непусто», а также в режиме «объединения», чтобы подсчитать больше фрагментов, но это не восполнило большую часть недостатка.
Столбцы 3 и 4 таблицы 2 показывают, что featureCounts работал примерно в пять раз быстрее и использовал примерно в 10 раз меньше памяти, чем следующий по эффективности инструмент.
7 ПРОИЗВОДИТЕЛЬНОСТЬ ПРИ БОЛЬШОМ КОЛИЧЕСТВЕ ЭТАЛОННЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
7.1 Симулированные данные
Наборы данных с большим количеством эталонных последовательностей представляют собой сложную задачу, поскольку программное обеспечение для подсчета прочтений должно эффективно сопоставлять имена контигов объектов с именами прочтений. Чтобы проверить производительность в этих условиях, мы смоделировали чтение из не полностью собранного генома с относительно большим количеством каркасов. Мы использовали сборку генома волнистого попугайчика, созданную в рамках проекта Assemblathon 2 (Bradnam et al., 2013; Ховард и др. , 2013). Для этой сборки имеется 16 204 аннотированных гена со 153 724 экзонами, расположенными на 2850 каркасах. Из аннотированных экзонных областей в собранных каркасах случайным образом было извлечено восемь миллионов одноконцевых прочтений длиной 100 п.н. Смоделированные чтения были введены в файл SAM. Информация о сопоставлении прочтений была заполнена в соответствии с местами, из которых были извлечены чтения.
Для этой сборки имеется 16 204 аннотированных гена со 153 724 экзонами, расположенными на 2850 каркасах. Из аннотированных экзонных областей в собранных каркасах случайным образом было извлечено восемь миллионов одноконцевых прочтений длиной 100 п.н. Смоделированные чтения были введены в файл SAM. Информация о сопоставлении прочтений была заполнена в соответствии с местами, из которых были извлечены чтения.
7.2 Сравнительные характеристики
Затем смоделированные чтения были суммированы на генном уровне.В таблице 3 сравниваются featureCounts с sumOverlaps и htseq-count для этого набора данных. Как видно из данных RNA-seq, sumoOverlaps дает те же подсчеты, что и featureCounts , тогда как htseq-count дает немного меньше. featureCounts сохранил свое преимущество в эффективности по сравнению с другими методами в этой оценке, увеличив свое преимущество в скорости по сравнению с summoverlaps в этом контексте.
Производительность при считывании РНК-секвенций, смоделированных на аннотированной сборке генома волнистого попугайчика
| Методы . | Количество чтений . | Время (мин) . | Память (МБ) . | |
|---|---|---|---|---|
| 9001 9002 | 7 924 065 | 9 924 0650,6 | 15 | 15 |
| Summarizeverllaps (весь генома сразу) | 7 924 065 | 12.6 | 2400 | |
| summarizeOverlaps (по помост) | 7 924 065 | 53,3 | 262 | |
| htseq-кол | 7 912 439 | 12,1 | 78 |
| Методы . | Количество чтений . | Время (мин) . | Память (МБ) . | |
|---|---|---|---|---|
| число функций | 7 924 065 | 0. 6 6 | 15 | |
| summarizeOverlaps (весь геном сразу) | 7 924 065 | 12,6 | 2400 | |
| summarizeOverlaps (по помост) | 7 924 065 | 53,3 | 262 | |
htseq-count | 7 912 439 | 9 912 439 | 12.1 | 78 | 78 |
Производительность с РНК-SEQ Читает, смоделированные из аннотированной сборки Genome Budgerigar
| Методы . | Количество чтений . | Время (мин) . | Память (МБ) . | ||
|---|---|---|---|---|---|
| featureCounts | 7 924 065 | 0,6 | 15 | ||
| summarizeOverlaps (весь геном сразу) | 7 924 065 | 12,6 | 2400 | ||
| summarizeOverlaps (на строительных лесах) | 7 924 065 | 53. 3 3 | 2 | 262 | |
| HTSEQ-Count 2 | 7 912 439 | 12.1 | 78 | 78 |
| Методы . | Количество чтений . | Время (мин) . | Память (МБ) . | |
|---|---|---|---|---|
| 9001 9002 | 7 924 065 | 9 924 0650,6 | 15 | 15 |
| Summarizeverllaps (весь генома сразу) | 7 924 065 | 12.6 | 2400 | |
| summarizeOverlaps (по помост) | 7 924 065 | 53,3 | 262 | |
| htseq-кол | 7 912 439 | 12,1 | 78 |
8 ТЕОРЕТИЧЕСКИЙ АНАЛИЗ АЛГОРИТМИЧЕСКОЙ СЛОЖНОСТИ
В этом разделе дается теоретический анализ времени вычислений и памяти, необходимых для featureCounts и других алгоритмов. Фактическое время и память, потребляемые компьютерной программой, зависят от компьютерного оборудования, операционной системы и других факторов, а также от математической эффективности используемого алгоритма. Однако мы можем вывести теоретические выражения для скорости, с которой время и память, используемые любым конкретным алгоритмом, должны увеличиваться с увеличением количества чтений, количества признаков и плотности признаков в геноме. Временная сложность алгоритма featureCounts может быть получена как , где f — количество признаков, r — количество прочтений и k 1 — количество признаков, включенных в геномный бин. .Это означает, что количество элементарных вычислений, используемых алгоритмом, увеличивается линейно с количеством чтений, независимо от количества признаков и несколько быстрее, чем линейно с количеством признаков. Пространственная сложность алгоритма featureCounts равна , что означает, что используемая память увеличивается линейно с увеличением количества функций плюс количество ячеек b 1 .
Фактическое время и память, потребляемые компьютерной программой, зависят от компьютерного оборудования, операционной системы и других факторов, а также от математической эффективности используемого алгоритма. Однако мы можем вывести теоретические выражения для скорости, с которой время и память, используемые любым конкретным алгоритмом, должны увеличиваться с увеличением количества чтений, количества признаков и плотности признаков в геноме. Временная сложность алгоритма featureCounts может быть получена как , где f — количество признаков, r — количество прочтений и k 1 — количество признаков, включенных в геномный бин. .Это означает, что количество элементарных вычислений, используемых алгоритмом, увеличивается линейно с количеством чтений, независимо от количества признаков и несколько быстрее, чем линейно с количеством признаков. Пространственная сложность алгоритма featureCounts равна , что означает, что используемая память увеличивается линейно с увеличением количества функций плюс количество ячеек b 1 . Временная и пространственная сложности для всех алгоритмов приведены в таблице 4.
Временная и пространственная сложности для всех алгоритмов приведены в таблице 4.
Теоретическая временная и пространственная сложность
Таблица 4.Теоретическая временная и пространственная сложность
Количество операций чтения обычно велико, поэтому скорость увеличения с r особенно важна. Алгоритм featureCounts имеет наименьшую временную сложность среди сравниваемых алгоритмов. Алгоритм поиска по красно-черному дереву, используемый htsesq-count , имеет более высокую сложность, поскольку обычно больше, чем квадратный корень из числа объектов на бин, используемый featureCounts .Иерархический поиск в бинах, используемых featureCounts , более эффективен, чем последовательный поиск, выполняемый coverageBED , потому что большинство операций чтения перекрывают несколько уровней бинов с coverageBED , в результате чего k 2 обычно больше, чем k 1 . countOverlaps и summoverlaps сортируют чтения в соответствии с их сопоставленными местоположениями, а затем используют дерево интервалов для поиска признаков, перекрывающихся с чтениями.Этап сортировки особенно дорог и вводит термины.
countOverlaps и summoverlaps сортируют чтения в соответствии с их сопоставленными местоположениями, а затем используют дерево интервалов для поиска признаков, перекрывающихся с чтениями.Этап сортировки особенно дорог и вводит термины.
Алгоритм htseq-count имеет наилучшую теоретическую пространственную сложность, но featureCounts не сильно отстает, потому что количество интервалов b 1 обычно мало по сравнению с f . BEDTols имеет более высокую пространственную сложность, чем featureCounts , поскольку использует больше бинов. CountOverlaps и summoverOverlaps имеют более высокую пространственную сложность, которая зависит от количества считываний, а также от количества признаков.
На практике время работы и использование памяти программным обеспечением определяются не только внутренней сложностью времени и пространства используемого алгоритма, но и эффективностью реализации программного обеспечения. Практические тайминги показывают, что featureCounts обеспечивает дополнительный прирост эффективности за счет высокопроизводительного программирования на C и прямого управления памятью.
Практические тайминги показывают, что featureCounts обеспечивает дополнительный прирост эффективности за счет высокопроизводительного программирования на C и прямого управления памятью.
9 ОБСУЖДЕНИЕ
Обобщение показаний — важный шаг во многих анализах данных секвенирования следующего поколения.В этом исследовании мы разработали новую программу обобщения прочитанного под названием featureCounts и сравнили ее с существующими методами с точки зрения эффективности и точности. Наши результаты показали высокую согласованность между альтернативными методами по точности суммирования. Однако наблюдалась большая разница в их вычислительной стоимости. Было обнаружено, что метод featureCounts в среднем на порядок быстрее и гораздо более эффективно использует память, чем другие методы. Высокая вычислительная эффективность featureCounts обусловлена его сверхбыстрым алгоритмом поиска признаков и его высокоэффективной реализацией, полностью использующей язык программирования C.
Все результаты, представленные в этой статье, были получены с использованием одного потока, но featuresCounts также поддерживает многопоточную обработку, что делает его особенно полезным для суммирования данных, полученных в больших исследованиях секвенирования. Это единственный существующий метод подсчета чтения, который поддерживает многопоточность.
Эта программа предоставляет широкий спектр опций, позволяющих пользователям полностью контролировать способ наилучшего суммирования прочитанных данных. Пользователи могут выбрать, должны ли они учитывать чтения, которые пересекаются с более чем одной функцией или мета-функцией.Этот выбор часто определяется типом эксперимента. Прочтения, перекрывающиеся с более чем одним геном (метапризнак), не должны учитываться в эксперименте с секвенированием РНК, потому что такие прочтения могут исходить только от одного гена, но обычно их следует учитывать в экспериментах с гДНК-секвенированием, таких как гистоновый чип. -последовательный эксперимент.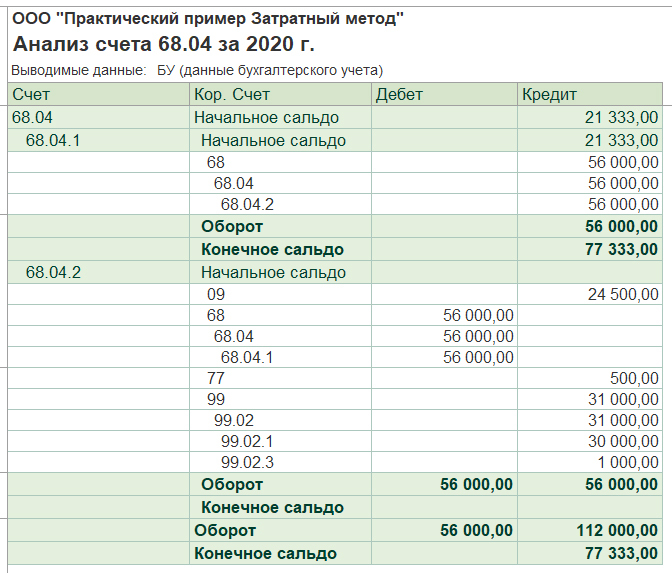 Эта программа также позволяет пользователям отфильтровывать чтения перед суммированием, используя ряд показателей, таких как показатели качества сопоставления, возможность сопоставления фрагментов (успешно сопоставлены два конца одного и того же фрагмента или нет), длина фрагмента, цепочка, химеризм и так далее.Он может автоматически обнаруживать ввод чтения формата SAM или BAM и сортировать чтения по имени, если парные чтения не находятся в последовательных позициях во входе. Это также позволяет пользователям указать, должны ли подсчитываться те операции чтения, о которых сообщалось более чем с одним расположением сопоставления (многосопоставление). Многие из этих полезных функций не поддерживаются другими программами.
Эта программа также позволяет пользователям отфильтровывать чтения перед суммированием, используя ряд показателей, таких как показатели качества сопоставления, возможность сопоставления фрагментов (успешно сопоставлены два конца одного и того же фрагмента или нет), длина фрагмента, цепочка, химеризм и так далее.Он может автоматически обнаруживать ввод чтения формата SAM или BAM и сортировать чтения по имени, если парные чтения не находятся в последовательных позициях во входе. Это также позволяет пользователям указать, должны ли подсчитываться те операции чтения, о которых сообщалось более чем с одним расположением сопоставления (многосопоставление). Многие из этих полезных функций не поддерживаются другими программами.
Программа featureCounts была реализована как в пакете SourceForge Subread (Liao and Shi, 2013), так и в пакете Bioconductor Rsubread (Shi and Liao, 2013a).Функция R предоставляет пользователям интерфейс R, чтобы они могли получить доступ к этой программе из знакомой им среды R. Он вызывает базовую скомпилированную программу C для выполнения всех операций суммирования чтения и, следовательно, имеет ту же скорость и использование памяти, что и пакет SourceForge Subread , полностью написанный на C. Реализация featureCounts в R позволяет необходимо создать полные конвейеры для анализа данных секвенирования следующего поколения с использованием программного обеспечения Bioconductor.Например, функции, включенные в пакеты Bioconductor Rsubread , limma и edgeR , можно использовать для выполнения полных анализов секвенирования РНК и ChIP-seq гистонов, начиная с картирования прочтений, суммирования прочтений и, наконец, анализа дифференциальной экспрессии или дифференциальный анализ модификаций гистонов. Мы считаем, что благодаря высокой эффективности и точности программа featureCounts станет полезным инструментом в наборе инструментов биоинформатики для анализа данных секвенирования следующего поколения.
Он вызывает базовую скомпилированную программу C для выполнения всех операций суммирования чтения и, следовательно, имеет ту же скорость и использование памяти, что и пакет SourceForge Subread , полностью написанный на C. Реализация featureCounts в R позволяет необходимо создать полные конвейеры для анализа данных секвенирования следующего поколения с использованием программного обеспечения Bioconductor.Например, функции, включенные в пакеты Bioconductor Rsubread , limma и edgeR , можно использовать для выполнения полных анализов секвенирования РНК и ChIP-seq гистонов, начиная с картирования прочтений, суммирования прочтений и, наконец, анализа дифференциальной экспрессии или дифференциальный анализ модификаций гистонов. Мы считаем, что благодаря высокой эффективности и точности программа featureCounts станет полезным инструментом в наборе инструментов биоинформатики для анализа данных секвенирования следующего поколения.
ПОДТВЕРЖДЕНИЕ
Авторы благодарят Леминга Ши и Чарльза Ванга за предоставление экспериментальных данных SEQC и Аарона Луна за полезные комментарии.
Финансирование : Грант на проект (1023454) и стипендия (для GKS) от Австралийского национального совета по здравоохранению и медицинским исследованиям (NHMRC). Поддержка операционной инфраструктуры правительства штата Виктория и NHMRC IRIIS правительства Австралии.
Конфликт интересов : не объявлено.
ССЫЛКИ
, и другие.GenomicRanges: представление и обработка геномных интервалов
,2013
.HTSeq: анализ высокопроизводительных данных секвенирования с помощью Python
,2013
, .Анализ дифференциальной экспрессии для данных подсчета последовательностей
,Genome Biol.
,2010
, том.11
стр.R106
, и др.Обнаружение дифференциального использования экзонов из данных секвенирования РНК
,Genome Res.
,2012
, том.22
(стр.2008
—2017
), и др.Анализ дифференциальной экспрессии данных секвенирования РНК на основе подсчета с использованием R и Bioconductor
, Nat. протокол
протокол
2013
, том.8
(стр.1765
—1786
), .Двухэтапная модель Пуассона для проверки данных секвенирования РНК
,Статистические приложения в генетике и молекулярной биологии
10
(стр.1
—26
), и др.Гидроксиметилирование всего генома, протестированное с помощью анализа help-gt, показывает перераспределение при раке
,Nucleic Acids Res.
,2013
, том.41
(стр.e157
—e157
), и др.Assemblathon 2: оценка de novo методов сборки генома у трех видов позвоночных
,Gigascience
,2013
, vol.2
стр.10
Brent Lab
GTF2.2: Формат аннотации гена
,2013
Сент-Луис
Вашингтонский университет
, и др.Ensembl 2012
,Nucleic Acids Res.
,2012
, том.40
(стр.D84
—D90
), и др.Инструменты для картирования данных высокопроизводительного секвенирования
,Биоинформатика
,2012
, vol.
28
(стр.3169
—3177
) и др.Bioconductor: открытая разработка программного обеспечения для вычислительной биологии и биоинформатики
,Genome Biol.
,2004
, том.5
стр.R80
, .baySeq: эмпирические байесовские методы определения дифференциальной экспрессии в данных подсчета последовательностей
11
стр.422
, и др.Сравнение основанных на секвенировании методов определения профиля метилирования ДНК и идентификации моноаллельных эпигенетических модификаций
,Nat. Биотехнолог.
,2010
, том.28
(стр.1097
—1105
), и др.De novo секвенирование с высоким охватом и аннотированные сборки генома волнистого попугайчика
Браузер генома человека в UCSC
,Genome Res.
,2002
, том.12
(стр.996
—1006
) и др.Сверхбыстрое и эффективное с точки зрения памяти выравнивание коротких последовательностей ДНК с геномом человека
, Genome Biol.
2009
, том.10
, и др.Вум! точные веса открывают инструменты анализа линейных моделей для подсчета прочтений РНК-секвенций
,2013
, .RSEM: точный количественный анализ транскриптов по данным RNA-Seq с эталонным геномом или без него
12
стр.323
, .Быстрое и точное выравнивание коротких прочтений с помощью преобразования Берроуза-Уилера
25
(стр.1754
—1760
) и др.Формат выравнивания/карты последовательностей и SAMtools
,Биоинформатика
,2009
, vol.25
(стр.2078
—2079
) и др.Нормализация, тестирование и оценка частоты ложных открытий для данных секвенирования РНК
13
(стр.523
—538
), .Пакет Subread: набор инструментов для обработки данных секвенирования нового поколения
,2013
, и др.Выравниватель субпрочтений: быстрое, точное и масштабируемое картирование прочтений с помощью начального числа и голосования
, Nucleic Acids Res.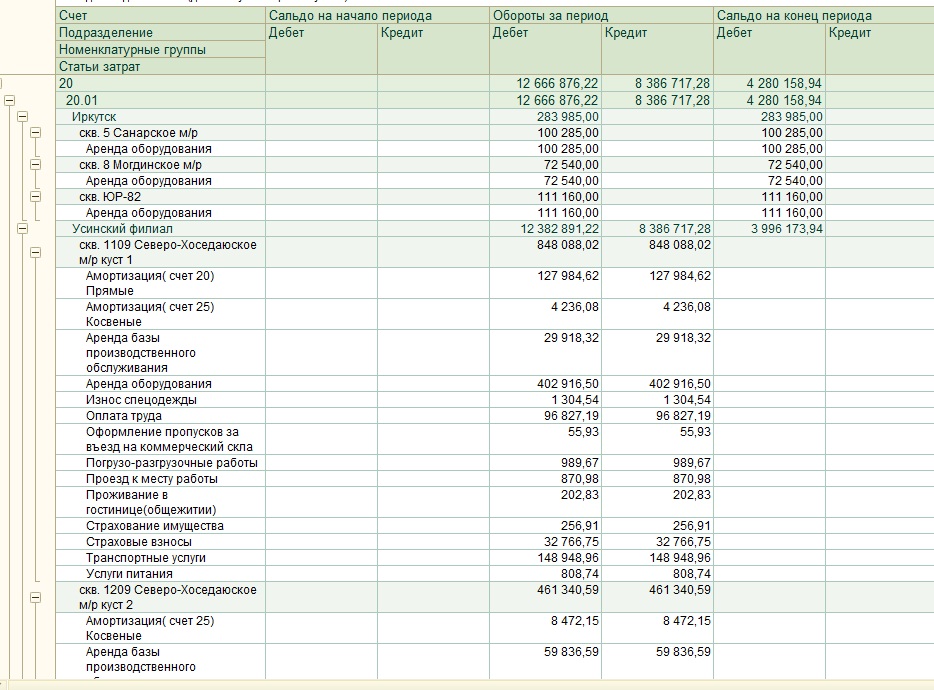
2013
, том.41
стр.e108
, и др.Фактор транскрипции IRF4 необходим для опосредованного аффинностью TCR метаболического программирования и клональной экспансии Т-клеток
,Nat. Иммунол.
,2013
, том.14
(стр.1155
—1165
) и др.Картограф GEM: быстрое, точное и универсальное выравнивание с помощью фильтрации
,Nat.Методы
,2012
, том.9
(стр.1185
—1188
) и др.Дифференциальный анализ экспрессии многофакторных экспериментов RNA-Seq в отношении биологической изменчивости
,Nucleic Acids Res.
,2012
, том.40
(стр.4288
—4297
).Технологии секвенирования нового поколения
,Nature Rev. Genet.
,2009
, том.11
(стр.31
—46
), и др.Всестороннее сравнение анализа транскриптома на основе RNA-Seq от прочтений до дифференциальной экспрессии генов и перекрестное сравнение с микрочипами: тематическое исследование Saccharomyces cerevisiae
, Nucleic Acids Res.
2012
, том.40
(стр.10084
—10097
), и др.От прочтений секвенирования РНК до результатов дифференциальной экспрессии
,Genome Biol.
,2010
, том.11
стр.220
, и др.IRanges: инфраструктура для управления интервалами в последовательностях
,, 2013,
, и др.Глобальные изменения в эпигеноме молочной железы индуцируются гормональными сигналами и координируются Ezh3
,Cell Rep.
,2013
, vol.3
(стр.411
—426
).Chip-seq: преимущества и проблемы развивающейся технологии
,Nat. Преподобный Жене.
,2009
, том.10
(стр.669
—680
) и др.Эталонные последовательности NCBI (RefSeq): текущий статус, новые функции и политика аннотации генома
,Nucleic Acids Res.
,2012
, том.40
(стр.D130
—D135
), .BEDTools: гибкий набор утилит для сравнения геномных признаков
,Биоинформатика
,2010
, том.
26
(стр.841
—842
) и др.Комплексная оценка методов дифференциального анализа экспрессии генов для данных Rna-seq
,Genome Biol.
,2013
, том.14
стр.R95
, и др.Дрейф и сохранение дифференциального использования экзонов в тканях у видов приматов
,Proc. Натл акад. науч. США
,2013
, том.110
(стр.15377
—15382
) и др.edgeR: пакет Bioconductor для дифференциального анализа экспрессии цифровых данных экспрессии генов
,Биоинформатика
,2010
, том.26
(стр.139
—140
), и др.Дифференциальное связывание рецепторов эстрогена связано с клиническим исходом рака молочной железы
,Nature
,2012
, vol.481
(стр.389
—393
).Секвенирование следующего поколения меняет современную биологию
,Nat. Методы
,, 2008,
, том.5
(стр.
16
—18
), .Rsubread: пакет R для выравнивания, обобщения и анализа данных секвенирования следующего поколения
,2013a
, .Subread/Rsubread Руководство пользователя
,2013b
, et al.Проект контроля качества микрочипов (MAQC) демонстрирует межплатформенную и внутриплатформенную воспроизводимость измерений экспрессии генов
,Nat. Биотехнолог.
,2006
, том.24
(стр.1151
—1161
) и др.TopHat: обнаружение сплайс-соединений с помощью РНК-seq
,Биоинформатика
,2009
, том.25
(стр.1105
—1111
) и др.Сборка транскриптов и количественная оценка с помощью секвенирования РНК выявляют неаннотированные транскрипты и переключение изоформ во время дифференцировки клеток
,Nat. Биотехнолог.
,2010
, том.28
(стр.511
—515
) и др.Полногеномный анализ сайтов связывания факторов транскрипции на основе данных секвенирования чипов
, Nat. Методы
Методы
, 2008,
, том.5
(стр.829
—834
)Wellcome Trust Sanger Institute
GFF (General Feature Format) технические характеристики, документ
,2013
, и др.Новый оценщик усадки для дисперсии улучшает обнаружение дифференциальной экспрессии в данных секвенирования РНК.
14
(стр.232
—243
) и др.Модельный анализ ChIP-Seq (MACS)
,Genome Biol
,2008
, vol.9
стр.Р137
Примечания автора
© The Author, 2013. Опубликовано Oxford University Press.Все права защищены. Для разрешений, пожалуйста, по электронной почте: [email protected]
Antibody Capture и CRISPR Guide Capture Analysis — Программное обеспечение — Экспрессия гена одной клетки — Официальная поддержка 10x Genomics
Cell Ranger 6.1 (последняя версия), напечатано 30. 01.2022
01.2022
Содержание
Обзор
Cell Ranger обрабатывает все данные штрих-кодов функций через конвейер подсчета, который количественно оценивает каждую функцию в каждой ячейке. Этот анализ проводится счетчик сотовых рейнджеров конвейер.Конвейер выводит единый матрица штрих-кодов признаков , которая содержит количество экспрессий генов наряду с Штрих-код функции подсчитывается для каждого штрих-кода ячейки. Матрица характеристик штрих-кода заменяет матрицу генного штрих-кода, испускаемую более старыми версиями Cell Ranger.
Конвейер сначала извлекает и исправляет штрих-код ячейки и UMI из библиотека функций, использующая те же методы, что и обработка чтения экспрессии генов. Это затем сопоставляет считанный штрих-код функции со списком объявленных функций в Справочнике по штрих-кодам функций.Количество для каждой функции доступны в функция-штрих-код выходных файлах матрицы и в выходном файле браузера лупы.
Чтобы включить анализ штрих-кодов функций, подсчет сотовых рейнджеров требует двух новых входы:
- Библиотеки CSV передается на сотовый рейнджер с
--librariesи объявляет файлы FASTQ и тип библиотеки для каждого входной набор данных. В типичном анализе Feature Barcode будет два входных параметра.
библиотеки: одна для нормального считывания экспрессии одноклеточного гена, а другая для
Штрих-код функции читает.Этот аргумент заменяет аргумент
В типичном анализе Feature Barcode будет два входных параметра.
библиотеки: одна для нормального считывания экспрессии одноклеточного гена, а другая для
Штрих-код функции читает.Этот аргумент заменяет аргумент --fastqs. - Справочник по функциям CSV передается на сотовый рейнджер с
--feature-refфлаг и объявляет набор используемых реагентов Feature Barcode в эксперименте. Для каждого уникального штрих-кода функции этот файл объявляет имя и идентификатор функции, уникальная последовательность штрих-кода функции, связанная с с этим реагентом и шаблоном, указывающим, как извлечь штрих-код функции последовательность из последовательности чтения.См. Справочник по штрих-кодам функций для получения подробной информации о том, как построить ссылку на функцию.
 В типичном анализе Feature Barcode будет два входных параметра.
библиотеки: одна для нормального считывания экспрессии одноклеточного гена, а другая для
Штрих-код функции читает.Этот аргумент заменяет аргумент
В типичном анализе Feature Barcode будет два входных параметра.
библиотеки: одна для нормального считывания экспрессии одноклеточного гена, а другая для
Штрих-код функции читает.Этот аргумент заменяет аргумент После создания CSV-файлов и настройки кода, выделенного красным, запустите cellranger count :
cd /home/jdoe/runs
количество рейнджеров --id=sample345 \
--libraries=library. csv \
--transcriptome=/opt/refdata-gex-GRCh48-2020-A \
--feature-ref=feature_ref.csv \
--expect-cells=1000
 csv \
--transcriptome=/opt/refdata-gex-GRCh48-2020-A \
--feature-ref=feature_ref.csv \
--expect-cells=1000
csv \
--transcriptome=/opt/refdata-gex-GRCh48-2020-A \
--feature-ref=feature_ref.csv \
--expect-cells=1000
Полный набор аргументов для подсчета рейнджеров описан в Одновыборочный анализ.
Типы функций и библиотек
При вводе данных штрих-кода функции в Cell Ranger через CSV-файл библиотеки,
вы должны объявить library_type каждой библиотеки. Конкретные значения
для library_type включит дополнительную нисходящую обработку,
специально для CRISPR Guide Capture и Antibody Capture. Следующая таблица
описывает типы библиотек, которые могут быть указаны, и что они означают для
последующая обработка.
| library_type | Описание |
|---|---|
Antibody Capture | Для использования в экспериментах по измерению уровней экспрессии белков клеточной поверхности с помощью окрашивания антител и/или антиген-мультимера. Включает t-SNE-проекцию клеток, используя только подсчет функции «Захват антитела/белок клеточной поверхности». Эта проекция доступна в выходном файле и в браузере лупы. Подробнее см. на странице «Алгоритмы антител». Включает t-SNE-проекцию клеток, используя только подсчет функции «Захват антитела/белок клеточной поверхности». Эта проекция доступна в выходном файле и в браузере лупы. Подробнее см. на странице «Алгоритмы антител». |
CRISPR Guide Capture | Позволяет анализировать изменения экспрессии генов, вызванные наличием возмущений CRISPR, в анализе типа Perturb-Seq. Дополнительные сведения см. на странице обзора CRISPR. В этом режиме также создается проекция t-SNE с использованием только направляющих отсчетов CRISPR.Эта проекция доступна в выходном файле и в браузере лупы. |
Пользовательский | Обеспечивает обработку считываний штрих-кода функции и базовую сводку качества секвенирования и качества библиотеки, но не выполняет специальной обработки счетчиков штрих-кода функции. |
Библиотеки CSV-файл
Файл CSV «Библиотеки» объявляет входные данные FASTQ для библиотек, создающих
провести эксперимент со штрих-кодом. Это будет включать одну библиотеку, содержащую Single
Считывания экспрессии гена клетки и одна или несколько библиотек, содержащих штрих-код признаков
читает.Чтобы использовать Cellranger count в режиме Feature Barcode, вы должны
создайте CSV-файл библиотек и передайте его с флагом
Это будет включать одну библиотеку, содержащую Single
Считывания экспрессии гена клетки и одна или несколько библиотек, содержащих штрих-код признаков
читает.Чтобы использовать Cellranger count в режиме Feature Barcode, вы должны
создайте CSV-файл библиотек и передайте его с флагом --libraries . То
В следующей таблице описано, какое содержимое должно быть в CSV-файле библиотек.
| Имя столбца | Описание |
|---|---|
fastqs | Полный путь к каталогу демультиплексированных файлов FAST для демультиплексированных файлов FAST . Аналогично --fastqs arg to cellranger count .Это поле не принимает пути с разделителями-запятыми. Если у вас есть несколько наборов быстрых запросов для этой библиотеки, добавьте дополнительную строку и используйте то же значение library_type . |
образец | То же, что и --sample arg to Cellranger count . Имя образца, указанное в листе образцов bcl2fastq. Имя образца, указанное в листе образцов bcl2fastq. |
тип_библиотеки | Данные FASTQ будут интерпретироваться с использованием строк из справочного файла функций, у которых «тип_функции» соответствует этому тип_библиотеки .Это поле чувствительно к регистру и должно соответствовать допустимому типу библиотеки, как описано в разделе «Типы библиотек/компонентов». Должен быть Gene Expression для библиотек экспрессии генов. (Сюда входит экспрессия целевого гена.) Должен быть одним из Custom , Antibody Capture или CRISPR Guide Capture для библиотек функциональных штрих-кодов. |
| Примечание. Для каждого уникального идентификатора образца требуется отдельная строка в CSV-файле библиотеки. |
Примеры файлов CSV для библиотек
Экспрессия генов + библиотеки CRISPR. В этом примере мы демультиплексировали
данные секвенирования из двух библиотек с именами GEX_sample1 и CRISPR_sample1 на листе образцов bcl2fastq / mkfastq. Это сгенерировало два файла FASTQ с именами
Это сгенерировало два файла FASTQ с именами GEX_sample1_S0_L001_001.fastq.gz и CRISPR_sample1_S0_L001_001.fastq.gz в пути /opt/foo (обязательно используйте правильный полный путь к файлам FASTQ).
Мы передаем образцы имен и путей FASTQ в Cell
Ranger с соответствующими типами библиотек:
| fastqs | образец | library_type |
|---|---|---|
| / Opt / Foo / | GEX_sample1 | Gene Expression |
| / Opt / Foo / | CRISPR_sample1 | CRISPR Руководство Capture |
Экспрессия генов + библиотеки антител. В этом примере мы демультиплексировали
данные секвенирования из двух библиотек с именами GEX_sample2 и Ab_sample2 на
образец листа bcl2fastq / mkfastq. Это сгенерировало два файла FASTQ с именами GEX_sample2_S0_L001_001.fastq.gz и Ab_sample2_S0_L001_001. в
путь  fastq.gz
fastq.gz /opt/foo (обязательно используйте правильный полный путь к файлам FASTQ).
Мы передаем имена образцов FASTQ в Cell Ranger с
соответствующие типы библиотек:
| fastqs | образец | library_type |
|---|---|---|
| / Opt / Foo / | GEX_sample2 | Gene Expression |
| / Opt / Foo / | Ab_sample2 | Антитело Capture |
Использование нескольких типов библиотек
Если ваша схема анализа создает библиотеку, содержащую несколько library_types , например, если вы используете CRISPR Guide Capture и
функции захвата антител, вам нужно будет выбрать один library_type для библиотеки при вводе в Библиотеки
CSV-файл.Это обеспечит только один вид специализированного библиотечного анализа. К
получить несколько специализированных анализов, вам нужно будет запустить несколько Cell Ranger
раз, передавая разные значения library_type в файле Libraries CSV
файл.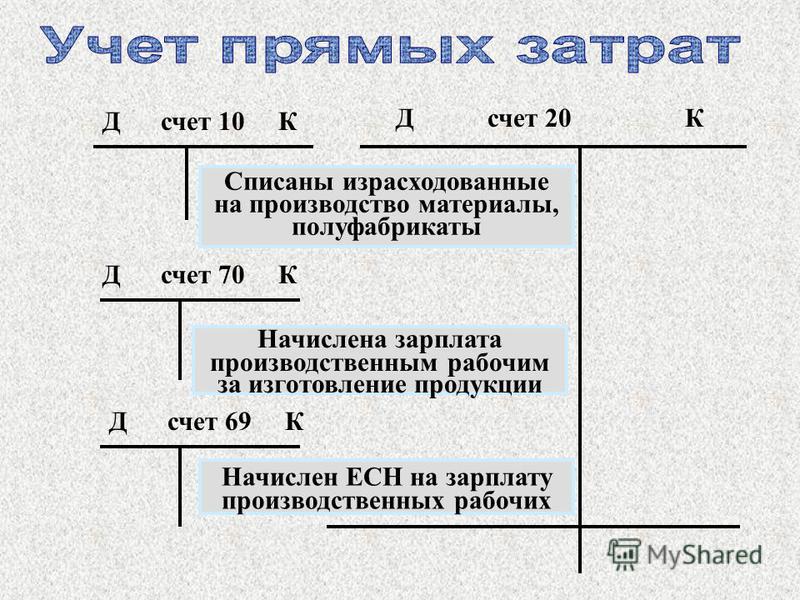 Это ограничение Cell Ranger, которое будет исправлено в будущем.
выпуск. Независимо от указанного
Это ограничение Cell Ranger, которое будет исправлено в будущем.
выпуск. Независимо от указанного library_type ,
Выходные данные матрицы штрих-кодов признаков будут содержать подсчеты для всех указанных признаков.
Справочник по функциям CSV-файл
Для обработки данных штрих-кода функции требуется CSV-файл с описанием функции.Он объявляет структуру молекулы и уникальную последовательность штрих-кода каждой характеристики.
особенность, присутствующая в вашем эксперименте. Каждая строка CSV объявляет один уникальный
Штрих-код функции. CSV-файл справки по функциям передается в cellranger.
count с флагом --feature-ref . Обратите внимание, что файл CSV
не может содержать символы вне диапазона ASCII.
Targeted Gene Expression совместимы с анализом Feature Barcode.
Однако, если данные экспрессии целевого гена анализируются в сочетании с
Данные штрих-кода функций на основе CRISPR, существуют дополнительные требования, предъявляемые к
CSV-файл справки по функциям. В частности, любые гены-мишени CRISPR направляющей РНК
(в столбце
В частности, любые гены-мишени CRISPR направляющей РНК
(в столбце target_gene_id CSV-файла справки по функциям)
должны соответствовать генам, которые также включены в экспрессию целевого гена
CSV-файл целевой панели (в столбце gene_id ).
В этой таблице описаны столбцы в CSV-файле справки по функциям. Пример файлы можно найти ниже.
| Имя столбца | Описание |
|---|---|
id | Уникальный идентификатор для этой функции.Не должно содержать пробелов, кавычек или запятых. Каждый идентификатор должен быть уникальным и не должен конфликтовать с идентификатором гена из транскриптома. |
имя | Удобочитаемое имя для этой функции. Не должно содержать пробелов. Это имя будет отображаться в браузере Loupe. |
чтение | Указывает, какое считывание последовательности РНК содержит последовательность Feature Barcode. Должен быть R1 или R2 .Примечание: в большинстве случаев R2 является правильным чтением. Должен быть R1 или R2 .Примечание: в большинстве случаев R2 является правильным чтением. |
узор | Указывает, как извлечь последовательность штрих-кода функции из прочитанного. Подробнее см. в разделе «Шаблон извлечения штрих-кода» ниже. |
последовательность | Последовательность штрих-кода нуклеотидов, связанная с этой функцией. Например, штрих-код антитела или последовательность протоспейсера sgRNA. |
тип_функции | Тип объекта.Подробнее о допустимых значениях этого поля см. в разделе «Типы библиотек/функций». Данные FASTQ, указанные в CSV-файле библиотеки с типом_библиотеки , совпадающим с типом_функции, будут сканироваться на наличие вхождений этой функции. Каждый тип функции в ссылке на функцию должен соответствовать записи library_type в файле Libraries CSV. Это поле чувствительно к регистру. |
target_gene_id | (необязательно) Идентификатор эталонного гена целевого гена направляющей РНК CRISPR.Ген с этим идентификатором должен существовать в эталонном транскриптоме. Предоставление target_gene_id и target_gene_name позволит конвейеру выполнять дифференциальный анализ выражений, предполагая, что контрольные («нецелевые») направляющие также указаны. Нецелевые руководства должны содержать значение «Нецелевые» в полях target_gene_id и target_gene_name . Дополнительные сведения см. в разделе «Обзор CRISPR». |
target_gene_name | (Необязательно) Имя гена целевого гена направляющей РНК CRISPR.Имя гена, соответствующее гену, указанному в поле target_gene_id , должно совпадать с именем гена, указанным здесь. Дополнительные сведения см. в разделе «Обзор CRISPR». |
Шаблон извлечения штрих-кода функции
Поле шаблона ссылки на функцию определяет, как найти
Штрих-код функции в пределах чтения. Штрих-код функции может появиться в известном месте.
смещение по отношению к началу или концу чтения или может появиться в фиксированном
положение относительно известной якорной последовательности.можно использовать вместо 5P ).
Штрих-код функции может появиться в известном месте.
смещение по отношению к началу или концу чтения или может появиться в фиксированном
положение относительно известной якорной последовательности.можно использовать вместо 5P ).
столбец последовательности ссылки на функцию.Должен появиться точно
один раз в узоре. Любые постоянные последовательности, состоящие из A , C , G и T в шаблоне
должны точно совпадать в последовательности чтения. Любой N в шаблоне может
соответствовать одному произвольному основанию. Для
свести к минимуму вероятность того, что ошибка последовательности сорвет матч. Фиксированный
последовательность также должна быть достаточно длинной, чтобы однозначно идентифицировать позицию
Штрих-код функции.Для типов объектов, которым требуется привязка, отличная от N, мы рекомендуем
12bp-20bp постоянной последовательности.
Любой N в шаблоне может
соответствовать одному произвольному основанию. Для
свести к минимуму вероятность того, что ошибка последовательности сорвет матч. Фиксированный
последовательность также должна быть достаточно длинной, чтобы однозначно идентифицировать позицию
Штрих-код функции.Для типов объектов, которым требуется привязка, отличная от N, мы рекомендуем
12bp-20bp постоянной последовательности.
Извлеченная последовательность штрих-кода объекта выравнивается со ссылкой на объект, и допускается не более одного базового несоответствия. Извлеченные последовательности штрих-кодов функций скорректировано до расстояния Хэмминга в одно основание с 10-кратной коррекцией штрих-кода Genomics алгоритм исправления штрих-кодов ячеек.
Захват антител с помощью TotalSeq™-B
TotalSeq™-B — это линейка
конъюгаты антитело-олигонуклеотид, поставляемые BioLegend, которые совместимы
с анализом Single Cell 3′ v3.Последовательность штрих-кода функции появляется в
фиксированная позиция (основание 10) в чтении R2.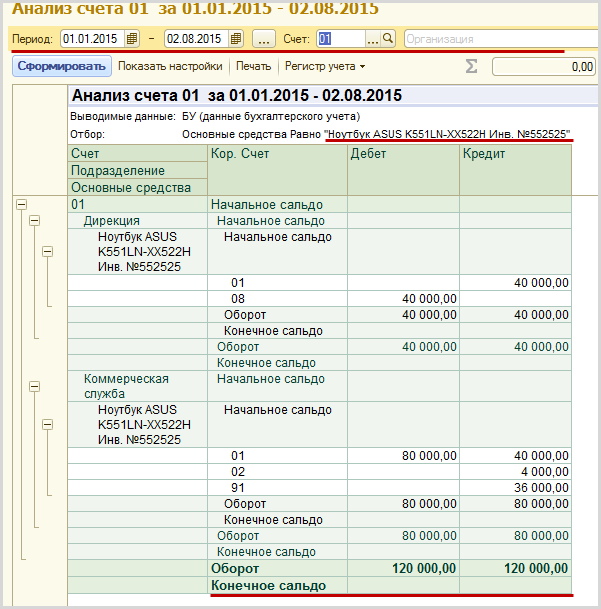
| чтение | узор |
|---|---|
| R2 | 5PNNNNNNNNN(BC) |
Пример набора данных
Пример TotalSeq™-B Feature Reference CSV Обратите внимание, что это предварительный набор антител TotalSeq-B. Особенность
С тех пор последовательности штрих-кодов изменились. Пожалуйста, обратитесь к
https://www.biolegend.com/totalseq для последнего сопряженного штрих-кода функции
Информация.
Захват антител с помощью TotalSeq™-C
TotalSeq™-C — это линейка конъюгаты антитело-олигонуклеотид, поставляемые BioLegend, которые совместимы с анализом Single Cell 5′. Последовательность штрих-кода функции появляется на фиксированной позиция (основание 10) в чтении R2.
| чтение | узор |
|---|---|
| R2 | 5PNNNNNNNNN(BC) |
Пример набора данных
Пример TotalSeq™-C Feature Reference CSV
Ссылка на функцию для
Библиотеки Immudex dMHC Dextramer® с декстрамерами dCODE
имеет тот же шаблон штрих-кода, что и TotalSeq™-C. Используйте «Захват антител» в
колонка
Используйте «Захват антител» в
колонка feature_type для декстрамерных или мультимерных реагентов. Следовательно
такой же
справочный пример функции для TotalSeq™-C
также может использоваться для библиотек MHC Dextramer®.
Захват антител для TotalSeq™-A
TotalSeq™-A — это линейка конъюгаты антитело-олигонуклеотид, поставляемые BioLegend, которые совместимы с комплектами Single Cell 3′ v2 и Single Cell 3′ v3. Штрих-код функции последовательность появляется в начале чтения R2.
Хотя TotalSeq™-A можно использовать с анализом CITE-Seq, CITE-Seq не является 10-кратным поддерживаемый анализ.Пожалуйста, свяжитесь с Нью-Йоркским центром генома или BioLegend за помощь в анализе или программное обеспечение.
Пример TotalSeq™-A Feature Reference CSV
CRISPR Guide Capture
В анализах CRISPR Guide Capture последовательность характеристического штрих-кода представляет собой
КРИСПР
последовательность протоспейсеров.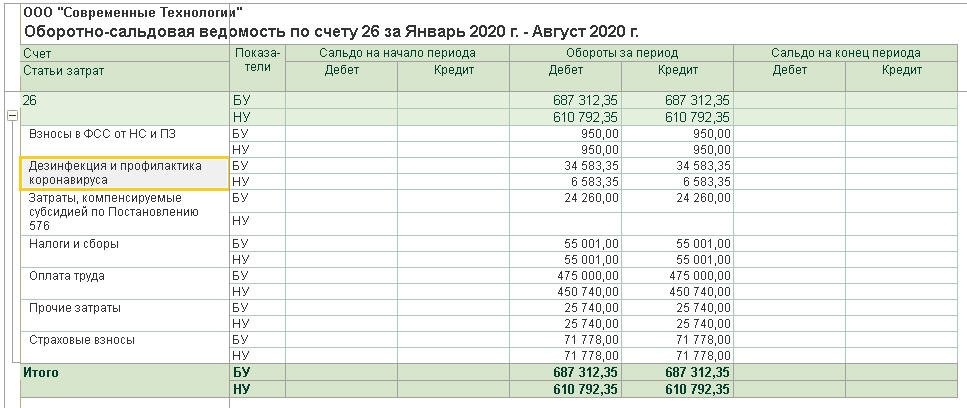 За протоспейсером следует нижележащая константа
последовательность в направляющей РНК, которая используется в качестве якоря для определения местоположения
протоспейсер. Мы рекомендуем использовать константную последовательность 12bp-20bp, которую можно
однозначно идентифицируется, но достаточно короток, чтобы его нельзя было нарушить
ошибка последовательности.В примере CSV-файла Feature Reference мы объявляем шесть
направлять функции РНК с помощью шести различных последовательностей штрих-кода / протоспейсера. Мы используем
За протоспейсером следует нижележащая константа
последовательность в направляющей РНК, которая используется в качестве якоря для определения местоположения
протоспейсер. Мы рекомендуем использовать константную последовательность 12bp-20bp, которую можно
однозначно идентифицируется, но достаточно короток, чтобы его нельзя было нарушить
ошибка последовательности.В примере CSV-файла Feature Reference мы объявляем шесть
направлять функции РНК с помощью шести различных последовательностей штрих-кода / протоспейсера. Мы используем столбцов target_gene_id и target_gene_name для объявления целевого гена
каждой направляющей РНК для использования в последующем анализе возмущений CRISPR. Два гида
объявлены с target_gene_id как Non-Targeting . Ячейки, содержащие Нецелевые направляющие будут использоваться в качестве средств контроля возмущения CRISPR.
анализ.Четыре оставшихся проводника нацелены на два гена.
| чтение | шаблон |
|---|---|
| R2 | (BC)GACCAGGATGGGCACCACCC |
Пример ссылки на функцию CRISPR CSV
Количество векторов в функции
Вектор для подсчета признаков
Используйте Vector to Feature Count для преобразования векторных записей в слой подсчета объектов.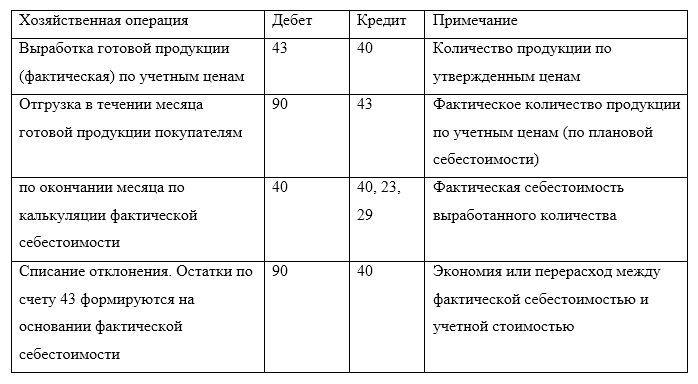
Вы также можете написать сценарий для преобразования векторных записей в слой подсчета объектов с помощью задачи VectorToFeatureCount.
Выполните следующие действия:
- На панели инструментов выберите Vector > Vector to Feature Count . Появится диалоговое окно «Преобразовать векторные записи в количество объектов».
- Укажите входной растр . Этот растр будет определять базовую проекцию для выходного слоя подсчета объектов.
- Укажите входной вектор . Это может быть любой векторный формат, поддерживаемый ENVI. Для полилиний и полигонов центр тяжести используется для создания точек подсчета объектов.
- Необязательно: В поле Имя атрибута столбца объектов укажите имя атрибута столбца, содержащего класс объектов.
- Необязательно: В поле Имя атрибута поля описания столбца укажите имя атрибута столбца, содержащего описание.
- Необязательно: В поле Имя атрибута столбца времени получения укажите имя атрибута столбца, содержащего время получения.
- В поле Output Feature Count укажите расположение и имя файла для выходного файла подсчета объектов (.efc). Расположением по умолчанию является каталог, указанный параметром Temporary Directory .
Чтобы запустить процесс на локальном или удаленном сервере ENVI, щелкните стрелку вниз и выберите Запустить задачу в фоновом режиме или Запустить задачу на Имя удаленного сервера ENVI .Консоль заданий ENVI Server покажет ход выполнения задания и предоставит ссылку для отображения результата после завершения обработки. Дополнительную информацию см. в разделе Серверы ENVI.
- Щелкните OK .
- По завершении обработки выберите Файл > Открыть в строке главного меню ENVI и выберите файл подсчета объектов.
- Когда в диалоговом окне Выбор данных будет предложено выбрать связанный растр для файла подсчета объектов, выберите тот же файл, который вы указали на шаге 2 выше.
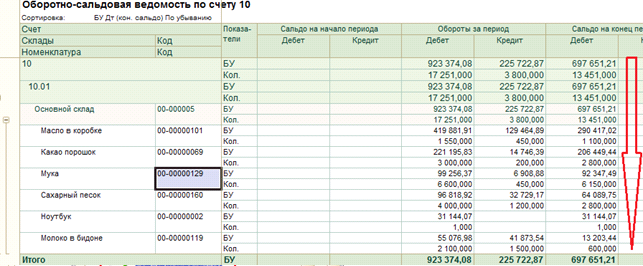

ENVI создает слой подсчета объектов и отображает его поверх входного растра. Он открывает инструмент подсчета объектов, чтобы вы могли просмотреть атрибуты слоя подсчета объектов.
Пример
В этом примере открывается шейп-файл городов США. Он группирует 3500 записей в 50 различных функций по штатам. Он создает файл подсчета функций ENVI (.efc), который вы можете просмотреть в Инструменте подсчета функций. Выполните следующие действия:
- На панели инструментов выберите Vector > Vector to Feature Count .Появится диалоговое окно «Преобразовать векторные записи в количество объектов».
- Нажмите кнопку Обзор рядом с полем Входной растр .
- Перейдите к папке данных в пути установки ENVI и выберите файл natural_earth_shaded_relief.jp2. Путь по умолчанию выглядит следующим образом, где xx — версия ENVI:
- Windows : C:\Program Files\Harris\ENVI xx \data
- Linux : /usr/local/harris/ENVI xx /данные
- Перейдите к папке classic\data\vector в пути установки ENVI и выберите файлы city.шп. Путь по умолчанию выглядит следующим образом, где xx — версия ENVI:
- Windows : C:\Program Files\Harris\ENVIxx\classic\data\vector
- Linux : /usr/local/harris/ENVI хх /классический/данные/вектор
- Этот шейп-файл содержит несколько столбцов атрибутов. Чтобы просмотреть их, щелкните правой кнопкой мыши citys.shp в диспетчере слоев и выберите View/Edit Attributes . Появится средство просмотра атрибутов:
- В поле Имя атрибута столбца признаков введите ST .
- В поле Имя атрибута столбца описания введите AREANAME .
- По умолчанию выходной файл подсчета объектов будет называться VectorToFeatureCount.efc. Он будет записан в каталог, указанный параметром Temporary Directory .Расположение по умолчанию:
- Windows : C:\Users\< имя пользователя >\AppData\Local\Temp
- Linux : /usr/local/tmp
- Нажмите OK .
- По завершении обработки выберите Файл > Открыть в строке главного меню ENVI.
- Перейдите во временный каталог и выберите VectorToFeatureCount.efc.
- Когда в диалоговом окне Выбор данных будет предложено выбрать связанный растр для файла подсчета объектов, выберите natural_earth_shaded_relief. jp2 и нажмите OK . ENVI создает слой подсчета объектов и отображает его поверх затененного рельефного изображения. Он открывает инструмент подсчета объектов, чтобы вы могли просмотреть атрибуты слоя подсчета объектов.
- Нажмите клавишу F12 на клавиатуре, чтобы увидеть заштрихованное рельефное изображение в полном объеме. Затем увеличьте масштаб США, чтобы увидеть количество функций; например:

В этом примере вы создадите 50 различных групп подсчета объектов на основе состояний, используя поле ST. Поле AREANAME будет использоваться для предоставления описания каждой записи подсчета объектов.
Поле AREANAME будет использоваться для предоставления описания каждой записи подсчета объектов.
 jp2 и нажмите OK . ENVI создает слой подсчета объектов и отображает его поверх затененного рельефного изображения. Он открывает инструмент подсчета объектов, чтобы вы могли просмотреть атрибуты слоя подсчета объектов.
jp2 и нажмите OK . ENVI создает слой подсчета объектов и отображает его поверх затененного рельефного изображения. Он открывает инструмент подсчета объектов, чтобы вы могли просмотреть атрибуты слоя подсчета объектов.Что мы выпустили: 20 новых функций и еще
Одной из ценностей, которой мы руководствуемся здесь, в Intercom, является доставка — это наше сердцебиение.Это воплощает нашу веру в то, что доставка — это только начало; что передача функций в руки клиентов — это самый быстрый способ получить обратную связь, чтобы мы могли продолжать итерацию.
Легко увлечься выпуском больших, блестящих новинок — в прошлом году мы запустили три из них (наши новые статьи о продуктах в базе знаний, наш новый Messenger и смарт-кампании), — но также есть большая ценность в выпуске регулярных, небольших улучшений и особенности тоже.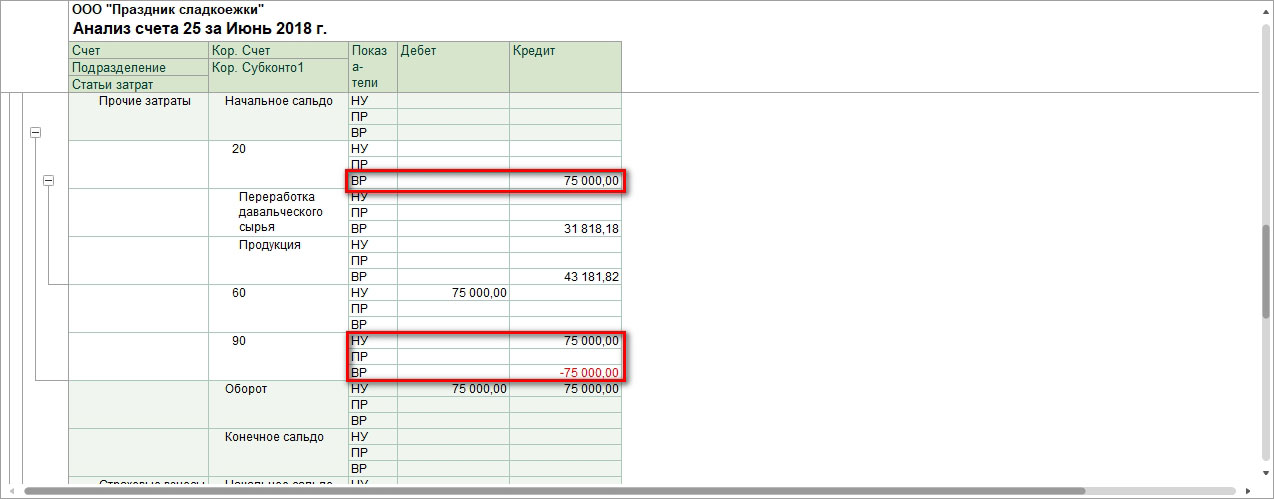 Они могут не оправдать громкое объявление, но эти небольшие улучшения могут оказать значительное влияние на наших клиентов.
Они могут не оправдать громкое объявление, но эти небольшие улучшения могут оказать значительное влияние на наших клиентов.
В этом году мы уже выпустили более 20 новых функций. Конечно, у нас все еще есть кое-что блестящее в рукаве, но вот некоторые из основных моментов того, что мы уже выпустили в 2017 году.
Изготовление опоры без трения и личной
Мы делаем все возможное, чтобы вам было проще предоставлять персональную и беспроблемную поддержку для ваших клиентов, поэтому мы выпустили ряд улучшений, которые помогут вам управлять ожиданиями ваших клиентов относительно того, когда они получат ответ, и сделать им легче найти помощь самим, если они не могут оставаться рядом.
Часы работы и режим отсутствия
Теперь вы можете лучше управлять ожиданиями клиентов с помощью рабочих часов и режима «отсутствия», которые позволяют вашим клиентам знать, когда ожидать ответа, и убедиться, что они не останутся без ответа.
Автоответчик статей
Позвольте клиентам быстрее получать ответы с помощью нашего автоответчика статей, который направляет их в ваш справочный центр, пока они ждут вашего ответа.
Мы также упростили процесс превращения Справочного центра статей в «вас», независимо от вашего размера, благодаря локализации на 38 языков и возможности добавить обратную ссылку на ваш веб-сайт.
Моар гифки
Ничто так не поднимает нам настроение, как своевременный GIF-файл, поэтому мы добавили поддержку анимированных GIF-файлов в Messenger, чтобы сделать ваши разговоры более увлекательными и индивидуальными.
Теперь также можно добавлять статьи, изображения, GIF-файлы и сохраненные ответы к сообщениям из наших мобильных приложений для Android и iOS, чтобы вы могли радовать клиентов даже в пути.
Помощь клиентам в масштабе
Мы постоянно работаем над тем, чтобы вам было проще масштабировать поддержку своих клиентов, чтобы по мере роста вашего бизнеса Интерком рос вместе с вами.
Мы добавили несколько новых функций, которые упрощают управление вашим почтовым ящиком и по-прежнему обеспечивают личную поддержку клиентов в больших командах:
Улучшения папки «Входящие»
Приоритизировать клиентов, которые дольше всего ждали ответа, на основе их первого сообщения с заказом «Длительнее всего ждал» в папке «Входящие» команды.
Групповые разговоры
Добавляйте товарищей по команде в беседы, чтобы вы могли общаться в группе, упрощая управление беседами и ускоряя их решение.
Отложить разговоры
Поддерживайте порядок в почтовом ящике, не теряя бесед, к которым хотите вернуться позже. Например, если вы хотите связаться с лидом, который не ответил в течение x дней, или вам нужно связаться с клиентом после дальнейшего изучения проблемы.
Облегчение поиска и создания интеграций
Мы знаем, насколько важно, чтобы Intercom хорошо взаимодействовал с другими инструментами, которые вы используете, поэтому мы продолжали фокусироваться на создании собственных интеграций, а также на упрощении работы других на нашей платформе.
Мы добавили Microsoft Teams в наш список интеграций, созданных Intercom, и продолжили расширять нашу программу Platform Partner, добавив несколько новых интеграций, а также упростив их обнаружение из нашего центра интеграции в приложениях.
Для тех, кто строит на нашей платформе, мы запустили новую панель инструментов разработчика, чтобы все, что вам нужно для разработки на основе API, было в одном доступном месте. Кроме того, мы добавили некоторые новые функции в наши API, такие как добавление потенциальных клиентов и компаний в наш API прокрутки.Подробнее о них можно прочитать в нашем блоге разработчиков.
Улучшение исходящих сообщений
Помимо оказания вам личной поддержки, мы также работали над тем, чтобы сделать проактивное общение с вашими клиентами — будь то регистрация, рекламные акции или объявления — более простым и целенаправленным:
Таргетинг на пользователей на основе взаимодействия
Теперь вы можете настроить таргетинг на пользователей в зависимости от того, насколько они вовлечены, с помощью фильтров, определяющих, открывали ли они ссылки в ваших предыдущих письмах или переходили по ним.
Останов ручных сообщений
Нажимайте кнопку «Отправить» в ручных сообщениях с большей уверенностью, так как теперь у вас есть возможность остановить сообщение после того, как оно было отправлено — для тех случаев, когда вы отправляете слишком рано или у вас есть сообщение, привязанное к времени.
Оптимизация для мобильной доставки
Убедитесь, что вы уведомляете мобильных пользователей в оптимальное время (а не посреди ночи) с новыми окнами доставки для push-уведомлений.
Как видите, в этом году мы были очень заняты, и впереди еще много работы.Нам не терпится поделиться с вами еще большим количеством вещей до конца года. Смотрите это пространство (или нашу страницу изменений!)
(PDF) Добыча добычи для локализованной толпы
2Chen, Loy, Gong, Xiang: добыча проекта для локализованной толпы Counting
Глобальная модель Наши Multi-
Модель
Несколько локальных
Модели
Глобальная функция Concateenated
Local Feature Local Feature
Global Ground
True
Local Ground
True
, которые можно сгруппировать, чтобы приблизить количество людей. Такой метод кластеризации движения, основанный на
работе, хорошо работает только при достаточно высокой частоте кадров видео, чтобы можно было надежно извлекать информацию о движении. Подсчет путем обнаружения и кластеризации основывается либо на
Подсчет путем обнаружения и кластеризации основывается либо на
, либо на явной сегментации объектов, либо на отслеживании характерных точек. Они не подходят для переполненных
сцен с захламленным фоном и частыми межобъектными перекрытиями. Напротив, подсчет
с помощью модели регрессии направлен на изучение прямого сопоставления между низкоуровневыми функциями и подсчетом людей
без разделения или отслеживания отдельных лиц.Этот подход больше подходит для
многолюдных сред и более эффективен в вычислительном отношении.
Существующие методы подсчета с помощью регрессионных методов можно разделить на глобальные подходы
или локальные подходы (см. рис. 1). Глобальные подходы [5,7,14,18,21] изучают единую функцию регрессии
между признаками изображения, извлеченными глобально из всего пространства изображения
, и общим количеством людей на этом изображении. Поскольку пространственная информация теряется при вычислении
глобальных признаков, такая модель неявно предполагает, что признак должен иметь один и тот же вес
независимо от того, где в сцене он извлечен. Однако это предположение в значительной степени неверно
Однако это предположение в значительной степени неверно
в реальных сценариях. В частности, структуры1 толпы могут различаться пространственно из-за плотности,
расположения сцены и самоорганизации толпы, вызванной элементарными индивидуальными взаимодействиями,
граничными условиями и правилами [11]. Таким образом, различные признаки могут быть более надежными и релевантными для подсчета толпы в разных местах. Кроме того, глобальная регрессионная модель не может предоставить информацию о пространственно-локальной информации о подсчете
толпы, которая требуется в некоторых приложениях.
Чтобы преодолеть эти ограничения глобального подхода, локальные модели [17,23] стремятся в определенной степени ослабить глобальное предположение, разделив пространство изображения на области ячеек, каждая из
которых моделируется отдельной функцией регрессии. Области могут быть ячейками, имеющими обычный размер
или различные разрешения, определяемые перспективой сцены для компенсации геометрических искажений камеры
[17]. Локальные подсчеты могут быть оценены в каждом регионе, а затем общий подсчет
Локальные подсчеты могут быть оценены в каждом регионе, а затем общий подсчет
может быть получен путем суммирования подсчетов на уровне ячеек.В крайнем случае Лемпицки и др.
др.[15] сделайте еще один шаг, чтобы смоделировать плотность толпы в каждом пикселе, представив задачу как
оценки плотности изображения, интеграл которой по любой области изображения дает количество
объектов в этой области. В целом, в отличие от глобальных подходов [5,7,14,18], локальные модели
нацелены на то, чтобы по-разному взвешивать признаки в зависимости от локальных структур толпы, чтобы облегчить локализованный
подсчет толпы. Однако существующие локальные методы страдают проблемой масштабируемости из-за необходимости изучения нескольких регрессионных моделей, число которых может стать очень большим.Кроме того,
неотъемлемым недостатком существующих локальных моделей является то, что информация не распределяется между пространственно локализованными
регионами, чтобы обеспечить более
точный подсчет толпы с учетом контекста. Во многих реальных случаях низкоуровневые объекты изображений могут быть очень
Во многих реальных случаях низкоуровневые объекты изображений могут быть очень
неоднозначными из-за загроможденного фона и серьезных перекрытий между объектами. Поэтому ха-
1Систематическое зернистое движение толпы, напоминающее течение газа, жидкости и сыпучих сред.
Список функций с принадлежащими токенами (доступное количество)
Список функций с принадлежащими токенами является необязательным и должен быть после заголовка журнала отчета. В этом разделе вы можете определить количество принадлежащих токенов (доступное количество) для функций в разделе «Список функций». Первая строка заголовка должна быть -Начало списка функций-, а последняя строка должна быть -Конец списка функций-.
В разделе «Список функций» каждая функция должна быть отформатирована следующим образом:
[название функции][разделитель][версия функции][разделитель][принадлежащие токены]
Разделитель задан в определении журнала отчета.
Примечание. Если журнал отчета не содержит раздела «Список функций» или если функция не указана в этом разделе, FlexNet Manager for Engineering Applications использует неограниченное количество токенов (доступное количество).
Пример списка функций
-Начало списка функций-
ф1,2.8,10
f2,2.8,8
ф3,1.0,20
-Конец списка функций-
См. также
Общие определения журнала отчетов
.id-o_1dp31c1ni102f1fp26l41cd0msrd.png) Online_Only__HelpNet___Local_Help_»> Форматирование общих файлов журнала отчетов
Online_Only__HelpNet___Local_Help_»> Форматирование общих файлов журнала отчетовОбщие примеры журналов отчетов
у.е.Отчет S. DOT «Каждое место имеет значение» включает скоростную автомагистраль Vine Street | Офис мэра | Пресс-релиз
В отчете Министерства транспорта США «Каждое место имеет значение» представлена скоростная автомагистраль Vine Street Expressway в Филадельфии
(Филадельфия, 20 декабря 2016 г.) — Усилия Филадельфии по улучшению скоростной автомагистрали Vine Street и прилегающих к ней районов освещаются в опубликованном отчете. сегодня Министерством транспорта США.
Министр транспорта США Энтони Фокс опубликовал краткий отчет «Лестницы возможностей, каждая точка имеет значение в дизайне». В отчете освещаются результаты двухдневных сессий по дизайну, проведенных ранее в этом году в Филадельфии, а также в Спокане, штат Вашингтон; Округ Рэмси/Хеннепин, Миннесота, и Нэшвилл, Теннесси.
В отчете освещаются результаты двухдневных сессий по дизайну, проведенных ранее в этом году в Филадельфии, а также в Спокане, штат Вашингтон; Округ Рэмси/Хеннепин, Миннесота, и Нэшвилл, Теннесси.
По словам Фокса, цель конкурса заключалась в том, чтобы «повысить осведомленность о существующих барьерах транспортной инфраструктуры и найти инновационные решения для восстановления связи сообществ с рабочими местами, здравоохранением, образованием и другими основными услугами».
Участие Филадельфии в усилиях началось весной этого года, когда Управление транспорта и инфраструктуры управляющего директора (OTIS) и Департамент улиц подали заявку на «Переосмысление Vine Street.В июле группа представителей Министерства транспорта США и технических экспертов собрала городских властей, градостроителей, дизайнеров и местных заинтересованных лиц для двухдневного обсуждения. Участники рассмотрели концепции дизайна и рекомендации по политике, которые могли бы помочь сплотить разделенные сообщества по всей скоростной автомагистрали.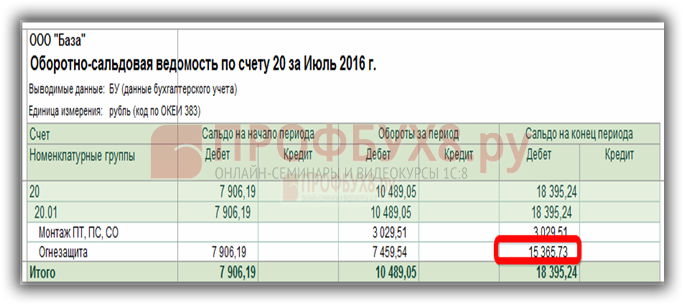
Переговоры привели к плану переосмысления коридора Вайн-стрит таким образом, чтобы улучшить связь между районами, создать равные возможности для многофункционального развития и инклюзивные варианты мобильности.Одним из приоритетов является обработка дорог и политические стратегии, которые повысят безопасность соединений и мобильность для всех. Городские власти и Комиссия по региональному планированию долины Делавэр взяли на себя обязательство совместно изучить, как можно изменить конфигурацию полос движения на Вайн-стрит, к востоку от Брод-стрит, чтобы снизить интенсивность движения и обеспечить более безопасные пешеходные переходы.
«Меня воодушевляет видение, созданное сообществом в результате этих усилий, — сказал мэр Кенни. «Я уверен, что та энергия, которую я увидел в той комнате, поможет воплотить рекомендации этого отчета в жизнь.Это огромное начало важной инициативы, и мы благодарим госсекретаря Фокса за его поддержку».
В отчете также определены две другие приоритетные области, такие как включение зеленой инфраструктуры, открытых пространств и ландшафта, а также поддержка роста сообщества и новых инвестиций для обеспечения равных и инклюзивных возможностей.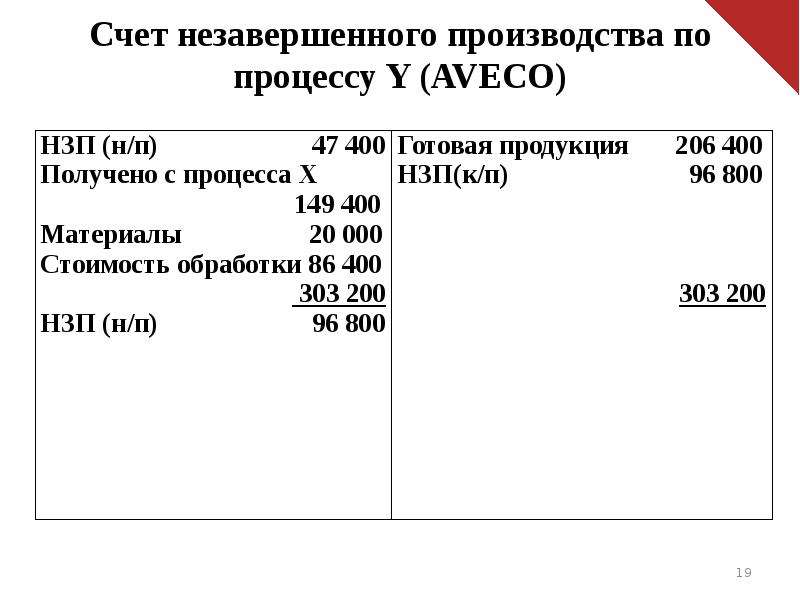 Ключевая местная заинтересованная сторона, Корпорация развития китайского квартала Филадельфии (PCDC), заинтересована в изучении этих приоритетов в рамках новых усилий по планированию района.
Ключевая местная заинтересованная сторона, Корпорация развития китайского квартала Филадельфии (PCDC), заинтересована в изучении этих приоритетов в рамках новых усилий по планированию района.
Джон Чин поддерживает инициативу: «Компания PCDC воодушевлена тем, что Филадельфия стала одним из четырех городов по всей стране, где обсуждались вопросы воссоединения сообществ, разделенных автомагистралями, и мы ценим региональные усилия, направленные на поддержку этой цели. После charrette PCDC начал процесс нового плана района, в котором Вайн-стрит была определена как основное направление».
Проект Design Challenge также положил начало важному диалогу между городскими властями и агентствами-партнерами.«Мы с нетерпением ожидаем совместной работы с городскими властями Филадельфии, DVPRC и Корпорацией развития китайского квартала Филадельфии над рекомендациями проектной сессии «Каждое место имеет значение», которые сделают районы рядом с коридором Вайн-стрит лучше для его жителей», — сказал помощник PennDOT.